Sådan fremstilles et lægemiddel. Trin 9: Indsendelse af godkendelsesansøgning til lægemiddelmyndighederne
Introduktion
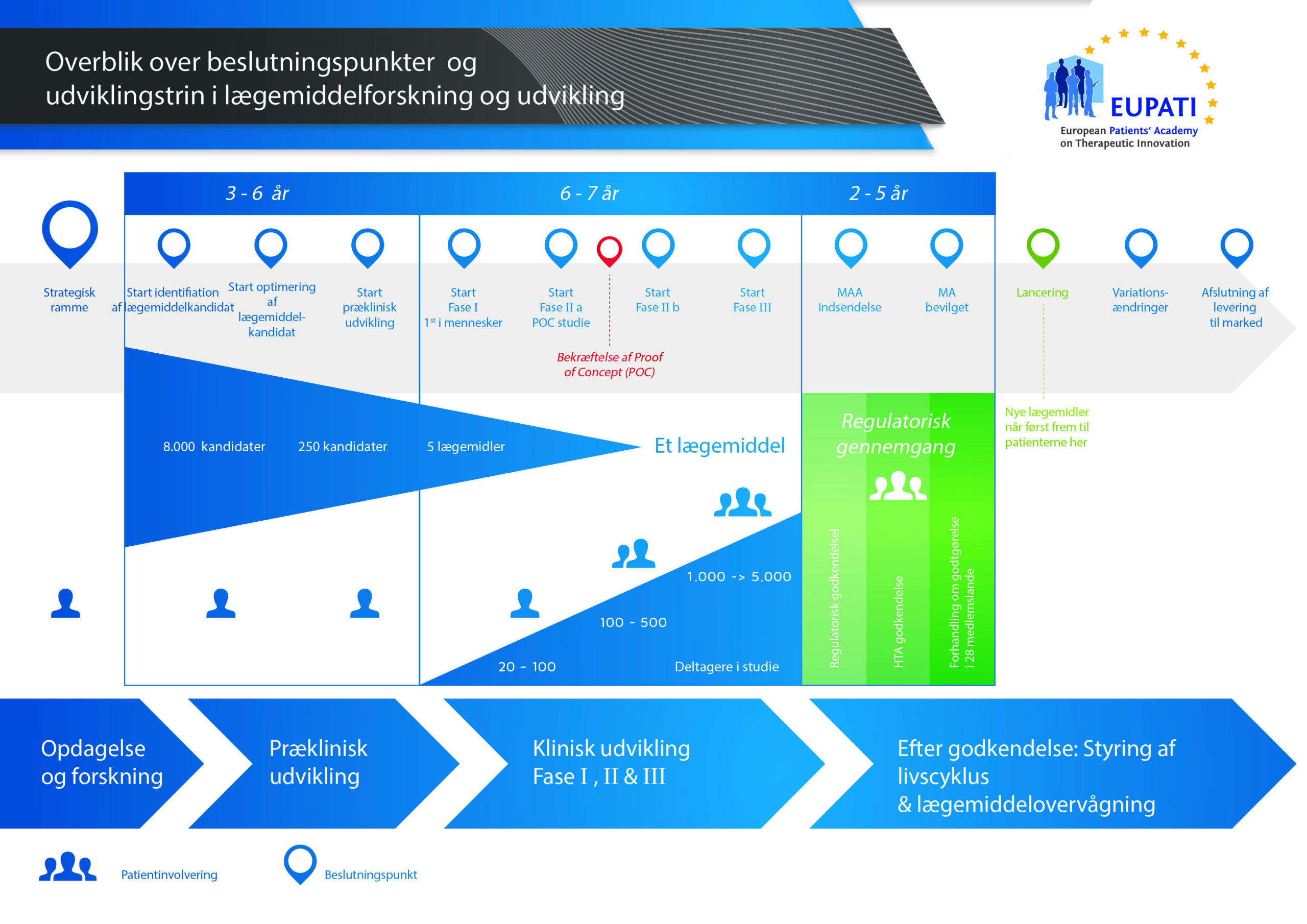
Det tager over 12 år og koster i gennemsnit over 1 milliarder kroner at gennemføre al den nødvendige forskning og udvikling, før et nyt lægemiddel er klar til brug for patienter.
Der er høj risiko forbundet med at udvikle lægemidler. De fleste stoffer (omkring 98 %), der udvikles, kommer aldrig på markedet som nye lægemidler. Det skyldes primært, at når man ser på fordelene og risiciene (de negative bivirkninger), i udviklingsfasen, kan de ikke matche de lægemidler, som patienterne allerede har adgang til.
Udviklingen af et nyt lægemiddel kan opdeles i 10 forskellige trin. Følgende artikel dækker trin 9: Indsendelse af godkendelsesansøgning til lægemiddelmyndighederne og ansøgning om markedsføringstilladelse.
#mla_gallery-1 { margin: auto; width: 100%; } #mla_gallery-1 .gallery-item { float: none; margin: 1.5%; display: inline-block; text-align: center; width: 97%; } #mla_gallery-1 .gallery-item .gallery-icon img { border: 2px solid #cfcfcf; } #mla_gallery-1 .gallery-caption { margin-left: 0; vertical-align: top; } /* see mla_gallery_shortcode() in media-library-assistant/includes/class-mla-shortcode-support.php */
-
- Det kræver langt over 10 års omhyggelig planlægning og forskning, før et lægemiddel kan tages fra molekyle til behandling, der er klar til at komme på markedet.
Trin 9: Indsendelse af godkendelsesansøgning til lægemiddelmyndighederne (ansøgning om markedsføringstilladelse)
Hvis resultaterne af de kliniske fase III-undersøgelser viser et acceptabelt forhold mellem fordele og risici, kan der udfærdiges en ansøgning om markedsføringstilladelse (MAA). Alle oplysningerne (prækliniske, kliniske og produktionsrelaterede) indsamles og organiseres i et forudbestemt format. Dette kaldes for en "dossier", og den sendes til lægemiddelmyndighederne. Personalets ekspertise i de afdelinger, der tager sig af regulatoriske forhold (farmaceutiske firmaer), er meget vigtig, da de forskellige lægemiddelmyndigheder over hele verden har lidt forskellige krav.
International Committee on Harmonisation (ICH) har harmoniseret mange af kravene i USA, Europa og Japan. Det har reduceret mængden af gentagne test og har forenklet processen, hvilket har resulteret i et fælles teknisk dokument (CTD – Common Technical Document), der kan gennemgås.
Når dossieren er modtaget, gennemgår lægemiddelmyndigheden oplysningerne og stiller spørgsmål, der skal besvares af personalet i den afdeling, der tager sig af regulatoriske forhold og har kontakt med lægemiddelmyndigheden, og som har indsendt dokumentet. Når lægemiddelmyndigheden er tilfreds med resultaterne (risici-fordele-vurdering), giver den sin godkendelse til, at det nye lægemiddel kan blive sendt på markedet. Gennemgangen tager normalt 12-18 måneder. Denne periode kan være kortere i særlige tilfælde, hvis dette er aftalt med lægemiddelmyndigheden, men den kan forlænges, hvis der er mange spørgsmål, der skal besvares. Myndighederne kan kræve flere kliniske undersøgelser, før de er klar til at give en godkendelse. Lægemidlet får ikke lov til at komme på markedet, før lægemiddelmyndigheden har godkendt det. Nogle gange er der omstændigheder, der ikke kan accepteres af lægemiddelmyndigheden, og lægemidlet bliver ikke godkendt til at komme på markedet.
I mange lande skal der også udføres undersøgelser om det nye lægemiddels omkostningseffektivitet. Disse dokumenter hjælper den offentlige myndighed eller forsikringsselskaberne via MTV-grupper (medicinsk teknologivurdering) med at tage stilling til og give anbefalinger om, hvorvidt lægemidlet skal ordineres og betales af forsikringssystemet i landet.
En kendt MTV-gruppe er National Institute for Clinical Excellence (NICE) i Storbritannien. NICE anbefaler, om den offentlige myndighed skal tillade, at lægemidlet ordineres.
Referencer
- Edwards, L., Fox, A., & Stonier, P. (Eds.). (2010). Principles and practice of pharmaceutical medicine (3rd ed.). Oxford: Wiley-Blackwell.
Bilag
A2-1.02.8-v1.1