Sådan fremstilles et lægemiddel. Trin 7: Fase II – bevis for effekt
Introduktion
Det tager over 12 år og koster i gennemsnit over 1 milliarder kroner at gennemføre al den nødvendige forskning og udvikling, før et nyt lægemiddel er klar til brug for patienter.
Der er høj risiko forbundet med at udvikle lægemidler. De fleste stoffer (omkring 98 %), der udvikles, kommer aldrig på markedet som nye lægemidler. Det skyldes primært, at når man ser på fordelene og risiciene (de negative bivirkninger), i udviklingsfasen, kan de ikke matche de lægemidler, som patienterne allerede har adgang til.
Udviklingen af et nyt lægemiddel kan opdeles i 10 forskellige trin. Følgende artikel dækker trin 7: Bevis for effekt – kliniske fase II-undersøgelser.
-
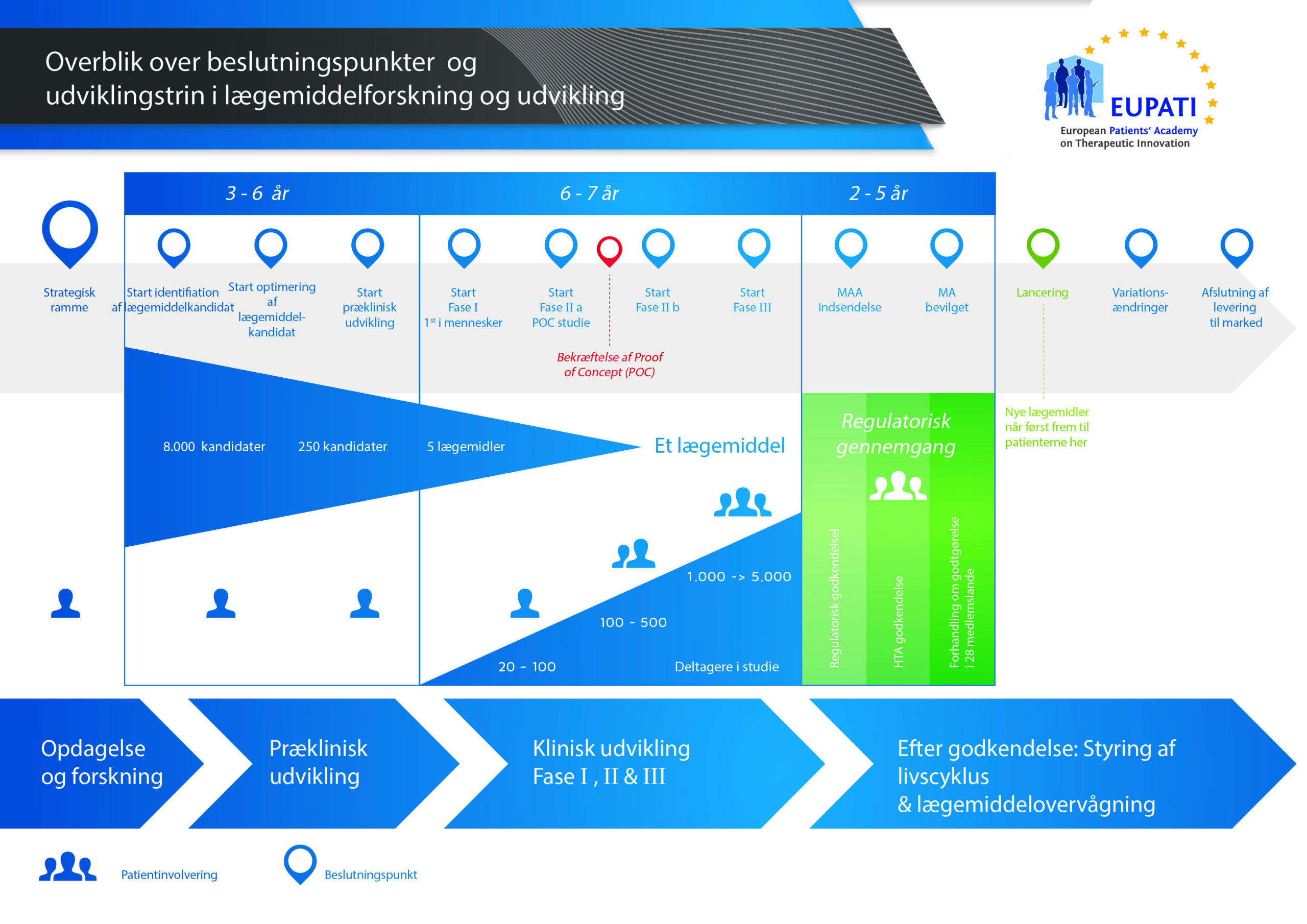
- Det kræver langt over 10 års omhyggelig planlægning og forskning, før et lægemiddel kan tages fra molekyle til behandling, der er klar til at komme på markedet.
Trin 7: Bevis for effekt – kliniske fase II-undersøgelser
Forsøg på patienter. Når resultaterne af undersøgelsen af raske frivillige har vist, at det er sikkert at gå videre, er næste skridt at starte kliniske forsøg på patienter med den sygdom, der behandles. For disse forsøg gælder de samme retningslinjer og bestemmelser som for fase I-forsøg.
I fase II- og fase III-undersøgelser er der normalt to behandlingsgrupper. Den ene gruppe har det aktive lægemiddel, og den anden gruppe for den nuværende bedste behandling eller et virkningsløst lægemiddel, der ikke har nogen effekt på kroppen (kaldet et "placebo"). Disse undersøgelser gennemføres normalt som "dobbeltblindede", "randomiserede" undersøgelser.
- "Dobbeltblindet" betyder, at både lægen og deltageren ikke er klar over, hvem der modtager det aktive lægemiddel eller den nuværende bedste behandling/placebo.
- "Randomiseret" betyder, at behandlingsgrupperne er tilfældigt udvalgt. Det gøres normalt med en computer, der genererer en tilfældig kode. Den kan ikke påvirkes af lægen eller nogen andre.
- "Placebokontrolleret" betyder, at nogle deltagere får et placebo, der gives på nøjagtig samme vilkår som det aktive lægemiddel. Det giver mulighed for at udskille de effekter, der er relateret til lægemidlet. Hvis en deltager i en undersøgelse f.eks. klager over hovedpine, er det vigtigt at vide, om det er relateret til det aktive lægemiddel. Hvis det samme antal deltagere, der får placebo, klager over hovedpine, viser det, at hovedpinen ikke kun skyldes det aktive lægemiddel.
Alle forsøgets detaljer beskrives i undersøgelsesprotokollen, og oplysningerne samles i en Case Record Form (CRF). Resultaterne analyseres herefter ved hjælp af statistiske test.
Disse forsøg udføres normalt på 100 til 500 patienter. De er designet til at indhente oplysninger om lægemidlets effekt på den aktuelle sygdom ("bevis for effekt"). Det er også i denne fase, at der bruges forskellige doser af lægemidlet for at finde ud af, hvad der er den bedste dosis. Denne dosis bruges herefter til den næste fase med større kliniske undersøgelser.
Jo mere der kan afdækkes om effekten i patienterne i denne fase, desto lettere er det at beslutte, om udviklingen af kandidatstoffet skal fortsætte. Fase II-undersøgelser er dog for små til at kunne levere tilstrækkelige beviser for virkningen og sikkerheden. Derfor er det vigtigt at indsamle mere og mere information om, hvordan lægemidlet virker i patienterne, for at reducere risikoen for, at det ikke består næste fase (fase III eller udvikling til lancering), som er den mest komplicerede og den dyreste udviklingsfase
Da disse fase II-undersøgelser udføres på patienter, gennemføres undersøgelserne normalt på flere forskellige hospitaler af hospitalslæger – også kaldet investigatorer – i modsætning til fase I-undersøgelser, som normalt gennemføres i særlige enheder.
Det er mere kompliceret at gennemføre forsøg på flere forskellige steder på samme tid, end det er at gennemføre et forsøg på et enkelt sted:
- Alle investigatorer og forsøgssygeplejersker skal være uddannet ved hjælp af en fastlagt protokol, så undersøgelsen udføres på samme måde på alle stederne.
- Lægemidlet skal eksporteres til andre lande og opbevares korrekt på forskellige apoteker.
- Blodprøver, der er taget fra patienterne i det kliniske forsøg, sendes normalt til et enkelt centralt laboratorium.
- Alle de lokale nationale regler og bestemmelser skal forstås og overholdes.
- Der er normalt krav om en godkendelse fra et etisk råd og den nationale kompetente myndighed i hvert land.
Alle disse aktiviteter skal koordineres af det globale undersøgelsesteam.
Sammenfatning: Trin 1-7
Ved slutningen af fase II-undersøgelser har programmet:
- i gennemsnit varet i 8,5 år og
- kostet ca. 7,5 milliarder kroner i gennemsnit.
For hver 10 lægemidler, der testes i fase I og fase II, er det kun to (i gennemsnit), der går videre til næste fase.
Referencer
- Edwards, L., Fox, A., & Stonier, P. (Eds.). (2010). Principles and practice of pharmaceutical medicine (3rd ed.). Oxford: Wiley-Blackwell.
Bilag
A2-1.02.6-v1.1