Sådan fremstilles et lægemiddel. Trin 6: Fase I – bevis for virkningsmekanisme
Introduktion
Det tager over 12 år og koster i gennemsnit over 1 milliarder kroner at gennemføre al den nødvendige forskning og udvikling, før et nyt lægemiddel er klar til brug for patienter.
Der er høj risiko forbundet med at udvikle lægemidler. De fleste stoffer (omkring 98 %), der udvikles, kommer aldrig på markedet som nye lægemidler. Det skyldes primært, at når man ser på fordelene og risiciene (de negative bivirkninger), i udviklingsfasen, kan de ikke matche de lægemidler, som patienterne allerede har adgang til.
Udviklingen af et nyt lægemiddel kan opdeles i 10 forskellige trin. Følgende artikel dækker trin 6: Bevis for virkningsmekanisme – kliniske fase I-undersøgelser.
-
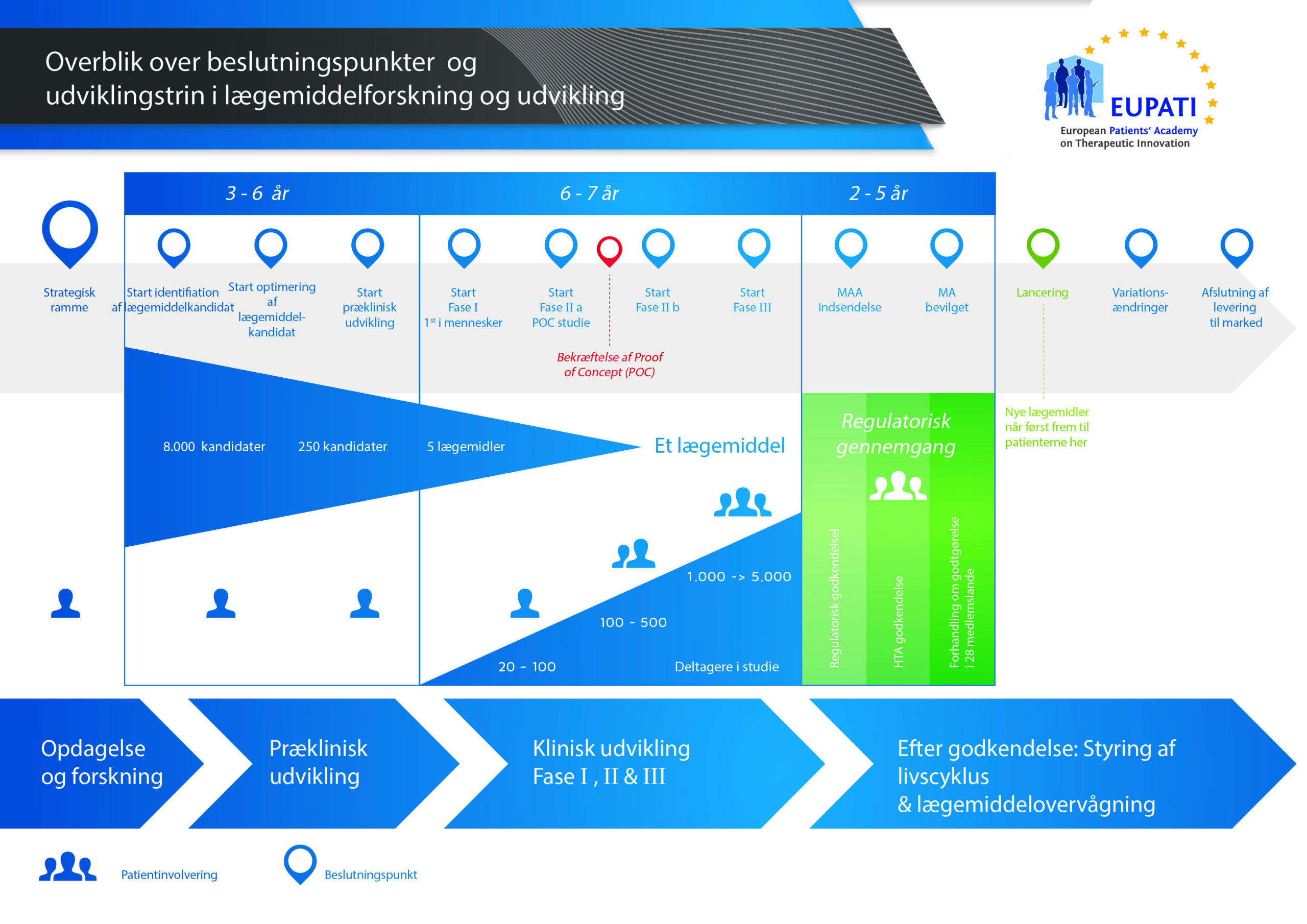
- Det kræver langt over 10 års omhyggelig planlægning og forskning, før et lægemiddel kan tages fra molekyle til behandling, der er klar til at komme på markedet.
Trin 6: Bevis for virkningsmekanisme – kliniske fase I-undersøgelser
Det er en stor beslutning at påbegynde den første kliniske undersøgelse. Når et kandidatstof fortsætter gennem udviklingsprocessen, stiger både antallet, omkostningerne og kompleksiteten af de aktiviteter, der er involveret i processen.
Før der påbegyndes en klinisk undersøgelse, skal der indsendes en ansøgning om et klinisk forsøg (Clinical Trial Application (CTA)). Ansøgningen om det kliniske forsøg skal indeholde følgende vigtige dokumenter:
- En Investigational Medicinal Product Dossier (IMPD), inklusive ADME og undersøgelser til at observere effekten (på forsøgspersonen), sikkerhedsoplysningerne om toksikologi samt oplysninger om, hvordan medicinen fremstilles.
- Undersøgelsesprotokollen, der beskriver detaljerne i forbindelse med udførelse af undersøgelsen og evaluering af resultaterne
- Investigator’s Brochure (IB), som indeholder et resumé af de oplysninger, som giver de læger, der tager sig af undersøgelsen (investigatorerne), en forståelse af, hvordan undersøgelsens medicin virker i kroppen (farmakologien). Det giver investigatorerne mulighed for at forklare undersøgelsen til den frivillige eller patienten og at indhente informeret samtykke (se nedenfor).
Ansøgningen om det kliniske forsøg skal sendes til godkendelse hos de kompetente nationale myndigheder (i Danmark er det Lægemiddelstyrelsen). I løbet af processen vil der også blive indhentet en vurdering fra den etiske komité.
Sikkerheden har topprioritet. Derfor kan en undersøgelse i mennesker ikke starte, før den interne kontrolkomité i virksomheden, den eksterne etiske komité og lægemiddelmyndighederne (Lægemiddelstyrelsen) har givet deres godkendelse.
Undersøgelser baseret på frivillige (også kaldet indledende undersøgelser, bevis for virkningsmekanisme-undersøgelser eller fase I-undersøgelser)
Denne undersøgelse giver læger og forskere mulighed for at kontrollere, om lægemidlet kan bruges sikkert i mennesker. Det kontrolleres også, om lægemidlet har samme virkning på mennesker, som det havde på dyr. Disse undersøgelser indeholder oplysninger om, hvordan lægemidlet virker – også kaldet "virkningsmekanisme". Disse undersøgelser har også til formål at opdage eventuelle sekundære virkninger af lægemidlet.
Cirka 20-100 frivillige er med i fase 1-kliniske undersøgelser. Disse undersøgelser udføres normalt i særlige fase 1-enheder, hvorfra de frivillige rekrutteres, og undersøgelserne gennemføres. De læger, der udfører disse undersøgelser, kaldes investigatorer. De er kvalificeret til at gennemføre kliniske forsøg med henblik på at fastslå resultatet af undersøgelsen.
Den første kliniske undersøgelse udføres normalt på raske mandlige frivillige. Detaljerne i den kliniske undersøgelse skal beskrives i undersøgelsesprotokollen og skal indeholde:
- baggrunden for sygdommen (det uopfyldte kliniske behov),
- de prækliniske oplysninger,
- detaljerne for den kliniske undersøgelse (hvad der præcis skal gøres og hvornår), og
- hvordan oplysningerne vil blive brugt og analyseret.
Alle de oplysninger, der kommer fra undersøgelsen, samles i et dokument, der hedder Case Record Form (CRF).
Her er der også et stort antal retningslinjer og bestemmelser, der er kendt som god klinisk praksis (GCP), for at beskytte deltagernes sikkerhed i undersøgelsen.
Undersøgelsesprotokollen har også et afsnit om "statistik", som er de statistiske test, der bruges til at analysere resultaterne. Der skal tages stilling til disse retningslinjer, før undersøgelsen starter, så det vides, hvordan oplysninger indsamles og anvendes, når undersøgelsen slutter.
To meget vigtige elementer er:
- Informeret samtykke (for at sikre, at deltagerne forstår, hvad det går ud på, og indvilger i at være med i undersøgelsen) og
- gennemgang og vurdering af den etiske komité.
Den etiske komité er en uafhængig gruppe, der normalt består af læger, videnskabsfolk, sygeplejersker og ikke-eksperter ("lægmænd"). De gennemgår undersøgelsesprotokollen (især formularen til informeret samtykke) og sikrer, at elementerne overholder komitéens etiske bestemmelser, før undersøgelsen udføres. Sikkerheden har højeste prioritet. For at sørge for deltagernes sikkerhed i en klinisk undersøgelse skal der foreligge en godkendelse internt i virksomheden, en positiv vurdering fra den eksterne etiske komité og godkendelse fra de kompetente nationale myndigheder (i Danmark Lægemiddelstyrelsen). Reglerne for fase I-undersøgelser er blevet endnu strengere efter et sjældent tilfælde i 2006, hvor frivillige oplevede alvorlige bivirkninger efter at have brugt et immunmodulerende lægemiddel til at behandle kronisk B-lymfocytær leukæmi og rheumatoid artrit.
Da sikkerheden har høj prioritet, lægger den første kliniske undersøgelse ud med en meget lav dosis af lægemidlet:
- Der bruges en enkelt dosis af lægemidlet til hver frivillig.
- Når det er blevet påvist, at der ikke er nogen sikkerhedsmæssige bekymringer med den første dosis, kan undersøgelsen fortsætte med en lidt højere dosis.
- Dosen øges derefter yderligere ("stigende dosis"), indtil den maksimalt tilladte dosis for undersøgelsen er nået.
Dette er beskrevet i undersøgelsesprotokollen.
Undersøgelsesresultaterne kan derefter analyseres, og alle sikkerhedsmålingerne kan vurderes. Det omfatter følgende:
- farmakokinetik – hvad kroppen gør ved lægemidlet. Blodniveauerne for lægemidlet kan måles – for at bestemme ADME (Absorption, Distribution, Metabolism og Excretion – absorbering, fordeling, metabolisme og udskilning)
- farmakodynamik – hvad lægemidlet gør ved kroppen ("virkningen"). Undersøgelsen kan f.eks. måle virkningen af et lægemiddel på visse blodceller.
Denne type undersøgelse er kendt som en SAD-undersøgelse (Single Ascending Dose – enkelt stigende dosis). Den efterfølges normalt af en MAD-undersøgelse (Multiple Ascending Dose – multipel stigende dosis), der involverer flere doser pr. frivillig, som navnet antyder.
Ud over SAD- og MAD-undersøgelserne skal der også udføres andre fase I-undersøgelser. F.eks.:
- for at undersøge indvirkningen af mad
- for at undersøge indvirkningen af andre lægemidler, der tages på samme tid
- for at undersøge indvirkningen af andre sygdomme, hvilket kan betyde, at der er behov for en anden dosis af lægemidlet (f.eks. i patienter med nyresygdomme).
Referencer
- Edwards, L., Fox, A., & Stonier, P. (Eds.). (2010). Principles and practice of pharmaceutical medicine (3rd ed.). Oxford, UK: Wiley-Blackwell.
Bilag
A2-1.02.5-v1.1