Realizzare un farmaco. Fase 9: presentazione della domanda di autorizzazione presso l’autorità di regolamentazione.
Introduzione
Sono necessari più di 12 anni e in media più di 1 miliardo di euro per condurre tutte le ricerche e lo sviluppo richiesti prima che un nuovo medicinale sia disponibile per l’uso da parte dei pazienti.
Lo sviluppo di farmaci è un’impresa ad alto rischio. La maggioranza (circa il 98%) delle sostanze sviluppate non riesce ad arrivare sul mercato come nuovo medicinale. Ciò accade soprattutto perché osservando i benefici e i rischi (effetti collaterali negativi) riscontrati durante lo sviluppo, non sono comparabili positivamente rispetto ai farmaci già disponibili per i pazienti.
Lo sviluppo di un nuovo farmaco può essere suddiviso in 10 fasi diverse. Il seguente articolo si occupa della Fase 9: presentazione della domanda e richiesta per l’ autorizzazione all’immissione in commercio presso l’autorità di regolamentazione
#mla_gallery-1 { margin: auto; width: 100%; } #mla_gallery-1 .gallery-item { float: none; margin: 1.5%; display: inline-block; text-align: center; width: 97%; } #mla_gallery-1 .gallery-item .gallery-icon img { border: 2px solid #cfcfcf; } #mla_gallery-1 .gallery-caption { margin-left: 0; vertical-align: top; } /* see mla_gallery_shortcode() in media-library-assistant/includes/class-mla-shortcode-support.php */
-
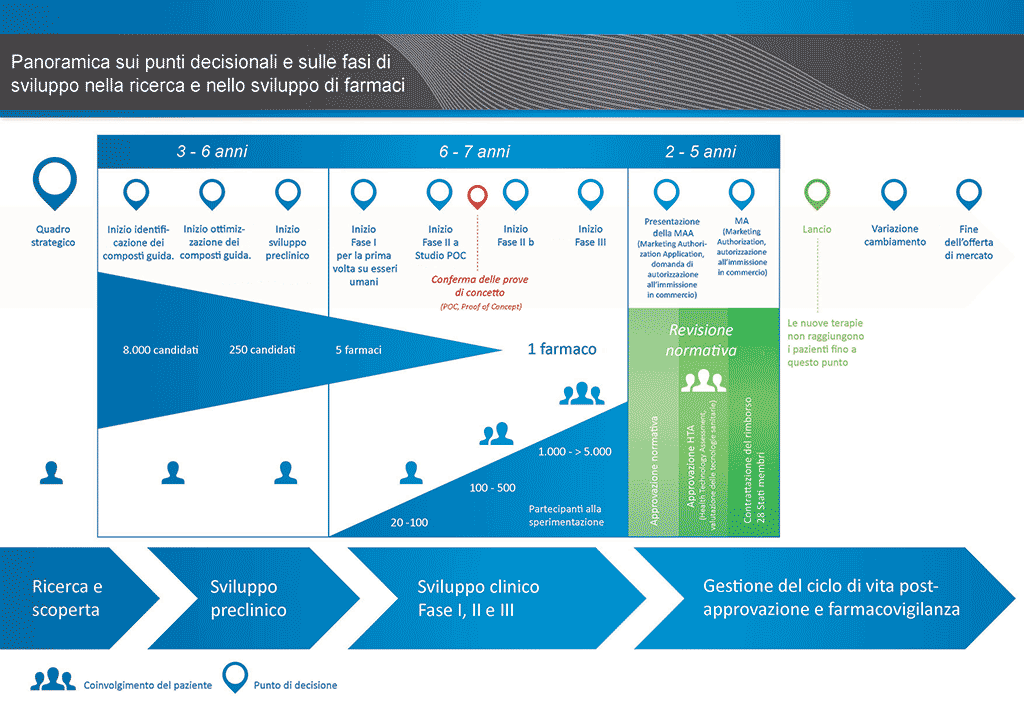
- Occorrono oltre 10 anni di attenta pianificazione e ricerca perché un farmaco passi da molecola a trattamento disponibile sul mercato.
Fase 9: Presentazione della domanda di autorizzazione all'immissione in commercio presso l'autorità di regolamentazione
Se i risultati degli studi clinici di Fase III mostrano un rapporto rischi benefici accettabile, è possibile preparare una richiesta per l'autorizzazione all'immissione in commercio (MAA, Marketing Authorisation Application). Tutte le informazioni (precliniche, cliniche e di fabbricazione) vengono raccolte e organizzate in un formato predefinito, Questa documentazione si chiama "dossier" ed è inviata alle autorità di regolamentazione. Le competenze del personale degli uffici per gli affari regolatori sono molto importanti poiché nel mondo le diverse autorità di regolamentazione hanno requisiti leggermente differenti l'una dall'altra.
La conferenza internazionale sull'armonizzazione (ICH, International Conference on Harmonisation) ha permesso di armonizzare molti dei requisiti per Stati Uniti, Europa e Giappone. In questo modo è stato possibile ridurre la duplicazione degli esami e semplificare il processo con il risultato di ottenere per la revisione un documento tecnico comune (CTD, Common Technical Document).
Una volta ricevuto il dossier, l'autorità di regolamentazione esaminerà le informazioni e presenterà delle domande a cui il personale dell'ufficio per gli affari regolatori che ha inviato il documento dovrà rispondere. Una volta che l'autorità di regolamentazione sarà soddisfatta dei risultati (valutazione dei rischi-benefici), essa darà la propria approvazione per l'immissione in commercio del nuovo medicinale. Il processo di revisione di solito richiede 12–18 mesi. Il periodo può ridursi in casi speciali se concordato da parte delle autorità di regolamentazione, ma può essere prolungato se vi sono molte domande a cui rispondere. Prima di essere disposte a dare la loro approvazione, le autorità possono richiedere ulteriori studi clinici e non sarà possibile immettere il farmaco sul mercato fino a che non saranno soddisfatte. A volte vi sono condizioni che le autorità di regolamentazione non possono accettare e l'immissione sul mercato del medicinale non sarà approvata.
In molti paesi, sono necessari anche studi sul rapporto costi-benefici del nuovo farmaco. Questi documenti aiuteranno le amministrazioni pubbliche o le aziende assicuratrici tramite i gruppi di valutazione delle tecnologie sanitarie (HTA, Health Technology Assessment) a decidere e a fornire raccomandazioni riguardo al permesso di prescrizione del medicinale e al suo finanziamento da parte del sistema assicurativo nazionale.
Un ente molto noto di valutazione delle tecnologie sanitarie è l'Istituto nazionale per l'eccellenza clinica (NICE, National Institute for Health and Care Excellence) nel Regno Unito. Il NICE fornisce all'amministrazione pubblica raccomandazioni relative al permesso di prescrivere i medicinali.
Riferimenti bibliografici
- Edwards, L., Fox, A., & Stonier, P. (Eds.). (2010). Principles and practice of pharmaceutical medicine (3rd ed.). Oxford: Wiley-Blackwell.
Allegati
#mla_gallery-2 { margin: auto; width: 100%; } #mla_gallery-2 .gallery-item { float: none; margin: 1.5%; display: inline-block; text-align: center; width: 97%; } #mla_gallery-2 .gallery-item .gallery-icon img { border: 2px solid #cfcfcf; } #mla_gallery-2 .gallery-caption { margin-left: 0; vertical-align: top; } /* see mla_gallery_shortcode() in media-library-assistant/includes/class-mla-shortcode-support.php */
- Scheda informativa: presentazione della domanda di autorizzazione presso l'autorità di regolamentazione.
Size: 97,419 bytes, Format: .docx
Questa scheda informativa descrive in dettaglio le attività relative alla presentazione del dossier finale sul farmaco alle autorità di regolamentazione, in preparazione della sua possibile immissione sul mercato.
#mla_gallery-3 { margin: auto; width: 100%; } #mla_gallery-3 .gallery-item { float: none; margin: 1.5%; display: inline-block; text-align: center; width: 97%; } #mla_gallery-3 .gallery-item .gallery-icon img { border: 2px solid #cfcfcf; } #mla_gallery-3 .gallery-caption { margin-left: 0; vertical-align: top; } /* see mla_gallery_shortcode() in media-library-assistant/includes/class-mla-shortcode-support.php */
- Presentazione: i principi di base relativi alla scoperta e allo sviluppo di farmaci
Size: 877,906 bytes, Format: .pptx
I principi di base relativi alla scoperta e allo sviluppo di farmaci. Sono necessari più di 12 anni e più di 1 miliardo di euro per condurre tutte le ricerche e lo sviluppo richiesti prima che un nuovo medicinale sia disponibile per l’uso da parte dei pazienti. Questa presentazione spiega in dettaglio il percorso a partire dalla scoperta fino all’immissione sul mercato di un nuovo farmaco e oltre.
A2-1.02.8-v1.1