Prognoseværdien af non-kliniske test
Introduktion
Helt fra meget tidligt i forsknings- og udviklingsprocessen for lægemidler er data, som indsamles i non-kliniske undersøgelser, afgørende for beslutninger om sikkerhed og virkning – f.eks. vedrørende planlægning af risikostyring, risikoreduktion, betingelser og specifikationer for markedsføringstilladelse, brugen af lægemidlet på markedet og sikkerhedsovervågning efter markedsføringen (lægemiddelovervågning).
Den information, der indsamles i non-kliniske undersøgelser, spiller en nøglerolle ved beslutninger:
- om kliniske forsøg
- om risikostyring og -reduktion
- om ansøgninger om markedsføringstilladelse
- om ordination af et lægemiddel til en patient
- om undersøgelser efter markedsføringen eller overvågningsundersøgelser
- og andet.
De følgende rammer viser de behov og faktorer, der er afgørende i forsknings- og udviklingsprocessen for lægemidler. Non-kliniske oplysninger spiller en nøglerolle ved fastlæggelsen af disse behov og faktorer. Denne artikel ser på non-kliniske undersøgelsers vigtige rolle som en betydende indikator for kliniske undersøgelser hos mennesker.
Nøglen til vellykket udvikling af lægemidler – retningslinjen de fem R’er1
- Rigtigt mål
- Stærk sammenhæng mellem lægemidlets mål og sygdommen
- Tilgængelige og prognostiske biomarkører
- Rigtigt væv
- Tilstrækkelig biotilgængelighed og vævseksponering
- Definition af farmakodynamiske biomarkører
- Klar forståelse af non-klinisk og klinisk farmakokinetik og farmakodynamik
- Forståelse af interaktioner med andre lægemidler (lægemiddelinteraktion)
- Rigtigt sikkerhed
- Klare sikkerhedsmargener
- Forståelse af sekundære farmakologiske risici
- Forståelse af aktive metabolitter, genotoksicitet og interaktioner med andre lægemidler
- Forståelse af farlige bivirkninger og andre ulemper
- Rigtige patienter
- Identifikation af den mest modtagelige patientpopulation
- Definition af benefit/risk-forholdet for den givne population
- Rigtiget kommercielle potentiale
- Cost/benefit i forhold til fremtidig standardbehandling
- Fokus på markedsadgang
Fra laboratorie- og dyreforsøg til patienter
Et kandidatstof kan ikke gives til mennesker, før der er indsamlet tilstrækkelige understøttende oplysninger om dets sikkerhedsprofil og de virkninger, det forventes at have. Non-kliniske undersøgelser tilvejebringer disse understøttende oplysninger via vigtige indikatorer som bevis for effekt (proof of concept), forslag til doseringsplan, tilstrækkelig farmakologisk kontrol og relevante in- og eksklusionskriterier.
Non-kliniske celleundersøgelser (in vitro) og dyreundersøgelser (in vivo) bør derfor:
- påvise virkningen af kandidatstoffet
- give viden om kandidatstoffets sikkerhedsprofil, f.eks. via undersøgelser af den maksimalt tolererede dosis
- vurdere effekter af kandidatstoffet, der ikke kan undersøges hos mennesker – f.eks. stoffets effekt på fosteret hos gravide kvinder.
Ekstrapolering fra dyr til mennesker
Ekstrapolering fra oplysninger, der er indsamlet om et lægemiddel i laboratoriet og ved dyrestudier, til anvendelse på mennesker kræver professionel vurdering. Der er udviklet og beskrevet nyttige regler for ekstrapoleringsprocesserne i retningslinjerne fra Det Europæiske Lægemiddelagenturs Udvalg for Humanmedicinske Lægemidler (CHMP)2 og det internationale råd for harmonisering (ICH)3. Retningslinjerne angiver de typer af undersøgelser, der skal foretages, inden der kan udføres kliniske forsøg.
Problemer med det non-kliniske program for et kandidatstof kan være årsag til indsigelser under evalueringen af ansøgningen om markedsføringstilladelse (MAA) i forbindelse med myndighedernes gennemgang. Sådanne sager kan føre til spørgsmål om relevansen af de non-kliniske modeller, der er brugt som basis for tilkendegivelsen om, at kandidatstoffet er beregnet til behandling af mennesker. For at sådanne problemer kan undgås, skal non-kliniske undersøgelser planlægges omhyggeligt, således at de forventninger, der er baseret på laboratorie- og dyrestudier, kan fungere som tilfredsstillende indikatorer.
Omfanget og området for det non-kliniske program, der skal være gennemført på tilfredsstillende vis, inden der kan indledes kliniske forsøg, varierer efter følgende faktorer:
- typen og sværhedsgraden af den sygdom, der er mål for behandlingen
- størrelsen af og dynamikken i den population, som kandidatstoffet skal behandle
- den kliniske forsøgsfase (fase I, II, III eller fase IV efter markedsføring)
- den forventede dosis og varighed af behandlingen hos mennesker.
Disse overvejelser bruges til at begrunde de typer af test eller dyremodeller, der bruges i det non-kliniske program.
Mange virksomheder søger råd om non-kliniske undersøgelser hos lægemiddelmyndighederne (f.eks. Det Europæiske Lægemiddelagentur (EMA) eller de kompetente nationale myndigheder). Disse videnskabelige råd hjælper virksomheden med at sikre, at de relevante test og undersøgelser gennemføres, så det ikke er sandsynligt, at der forekommer større indsigelser vedrørende testdesignet under behandlingen af MAA’en. Når virksomheden søger og følger myndighedernes råd, er der større sandsynlighed for et positivt resultat på MAA-stadiet. Rådgivningen sker i lyset af aktuel videnskabelig viden og er baseret på den dokumentation, som virksomheden leverer.
Non-kliniske data er vigtigst i de tidlige stadier af udviklingsprocessen for et kandidatstof (se figur 1). Når et lægemiddel er tilgængeligt til ordination (efter MAA), erstattes mange af de non-kliniske data om sikkerhed og virkning af data fra kliniske forsøg med patienter. Dog kan etiske overvejelser i nogle tilfælde hindre, at der indsamles data fra forsøgspersoner – f.eks. om effekten af et kandidatstof på udvikling af kræft eller på forplantning. I sådanne tilfælde styres den kliniske brug af nye lægemidler i længere tid af non-kliniske data. I sidste ende skal også disse data erstattes af data, der er indsamlet om sådanne forekomster, f.eks. som led i vedligeholdelse efter markedsføringen og lægemiddelovervågning.
-
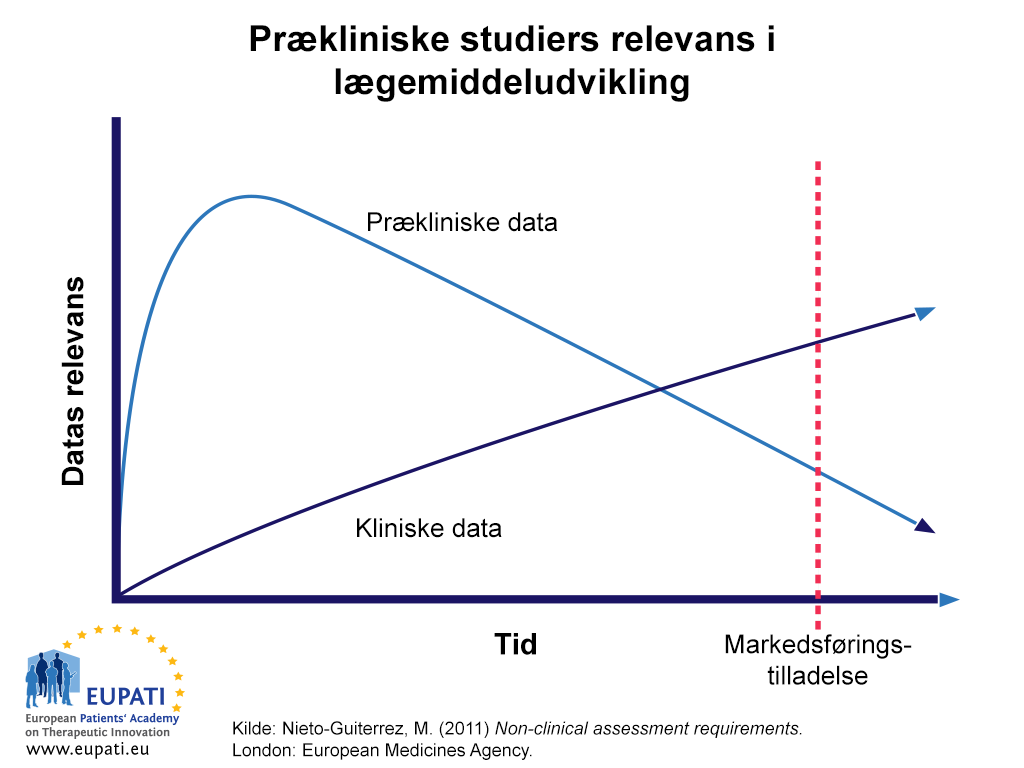
- Selvom prækliniske data er meget mere relevante tidligt i processen til udvikling af et lægemiddel, bliver deres relevans over tid overhalet af relevansen af kliniske data.
Figur 1 viser den relative vigtighed og afhængighed af non-kliniske data i udviklingsprocessen for lægemidler over tid. Data fra non-kliniske undersøgelser anvendes i større omfang end kliniske data indtil senere i udviklingsprocessen.
Ideelt set bør alle non-kliniske sikkerhedsproblemer, der fremkommer i udviklingsperioden, være løst på tidspunktet for ansøgningen om markedsføringstilladelse. Dog kan der på tidspunktet for indsendelse og vurdering af dossieret stadig være vigtige årsager til sikkerhedsproblemer, herunder f.eks. karcinogenicitet, genotoksicitet, genotoksiske urenheder, reproduktionstoksicitet og hepatotoksicitet.
Etiske overvejelser
Retningslinjer for, om brugen af dyr som modeller ved risikovurdering for mennesker og anvendelsen af disse modeller til at efterligne sygdomme hos mennesker er acceptabel, er fastlagt i Helsinki-deklarationen.4 Helsinki-deklarationen anfører den etiske og videnskabelige begrundelse for raske frivillige forsøgspersoners første eksponering for kandidatstoffer. Desuden fastlægger deklarationen, at biomedicinsk forskning bør være baseret på laboratorie- og dyreforsøg, der er gennemført i tilstrækkeligt omfang, samt på et indgående kendskab til den videnskabelige litteratur. Velfærden for dyr, der bruges til forskning, skal respekteres.
Non-kliniske undersøgelser: Tilstrækkelige indikatorer til undersøgelser hos mennesker?
Historisk set har udfordringer i forbindelse med non-kliniske undersøgelsers prognostiske værdi været forbundet med farmakokinetik, farmakodynamik (virkning) og sikkerhedsaspekter hos mennesker, som ikke umiddelbart kan forudsiges via non-kliniske undersøgelser. Mange nye in silico-teknologier (computermodeller), farmakogenomik, biomarkører og nye udforskningsdesign til kliniske forsøg er i hurtig udvikling, og de har alle en positiv indvirkning på non-kliniske undersøgelsers prognostiske værdi.
Flere ressourcer
- The Declaration of Helsinki is available in English, Spanish, and French at https://www.wma.net/policies-post/wma-declaration-of-helsinki-ethical-principles-for-medical-research-involving-human-subjects/ (Retrieved 4, July 2021). It is also available in Czech, German and Portuguese, at https://web.archive.org/web/20160517043747/http://www.wma.net/en/20activities/10ethics/10helsinki (Retrieved 4 July 2021)
Referencer
- Adapted from Cook, D., Brown, D., Alexander, R., March, R., Morgan, P., Satterthwaite, G., & Pangalos, M. (2014). Lessons learned from the fate of AstraZeneca's drug pipeline: A five-dimensional framework. Nature Reviews Drug Discovery, 13, 419-431. doi:10.1038/nrd4309
- European Medicines Agency. (2015) Non-clinical guidelines. Retrieved 24 July, 2015, from http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_000083.jsp&mid=WC0b01ac0580027548
- International Conference on Harmonisation (2015). ICH Guidelines. Retrieved 24 July, 2015, from http://www.ich.org/products/guidelines.html
- World Medical Association. (2013) WMA Declaration of Helsinki – Ethical Principles for Medical Research Involving Human Subjects. Retrieved 24 July, 2015, from http://www.wma.net/en/30publications/10policies/b3/
- Nieto-Guiterrez, M. (2011) Non-clinical Assessment Requirements. Brussels: European Medicines Agency. Retrieved 24 July, 2015, from http://www.ema.europa.eu/docs/en_GB/document_library/Presentation/2011/06/WC500107868.pdf
Bilag
A2-2.02.1-v1.3