Last update: 18 Luglio 2023


Principi sovrastanti al coinvolgimento dei pazienti nell’intero processo di ricerca e sviluppo dei farmaci
L’Accademia europea dei pazienti (EUPATI) è un progetto dell’Iniziativa paneuropea per i farmaci innovativi (Innovative Medicines Initiative, IMI) composto da 33 organizzazioni con partner che vanno da organizzazioni dei pazienti, università, organizzazioni no-profit ad aziende farmaceutiche. In EUPATI il termine “paziente” si riferisce a tutti i gruppi demografici in tutti i disturbi. EUPATI non si concentra su problemi o terapie specifici per una malattia, ma sul processo di sviluppo dei farmaci in generale. Informazioni relative a indicazioni, età o a trattamenti specifici con farmaci vanno al di là degli obiettivi di EUPATI e sono di competenza degli operatori sanitari nonché delle organizzazioni dei pazienti. Per approfondire visita eupati.eu/.
La grande maggioranza degli esperti coinvolti nello sviluppo e nella valutazione dei farmaci sono scienziati che lavorano sia nel settore privato che pubblico. Vi è un bisogno crescente di attingere alle conoscenze e alle esperienze dei pazienti al fine di comprendere cosa significa vivere con un particolare disturbo, in che modo la terapia viene somministrata e l’uso quotidiano dei farmaci. Queste opinioni contribuiscono a migliorare la scoperta, lo sviluppo e la valutazione di nuovi farmaci efficaci.
L’interazione strutturata tra pazienti di tutti i gruppi di età e condizioni, i loro rappresentanti e altre parti in causa è necessaria e consente lo scambio d’informazioni e un dialogo costruttivo a livello nazionale ed europeo dove i punti di vista degli utilizzatori dei farmaci possono e devono essere presi in considerazione. È importante prendere in considerazione che i sistemi sanitari nonché le pratiche e le normative possono differire.
Raccomandiamo la cooperazione e la partnership stretta tra le varie parti in causa incluse le organizzazioni degli operatori sanitari, organizzazioni di ricerca a contratto, organizzazioni di pazienti e di consumatori*, accademia, società scientifiche e accademiche, enti di regolamentazione, organismi di valutazione delle tecnologie sanitarie (health technology assessment, HTA) e l’industria farmaceutica. L’esperienza ad oggi dimostra che il coinvolgimento dei pazienti ha avuto come esito una maggiore trasparenza, fiducia e rispetto reciproco tra loro e altri portatori d’interesse.
È riconosciuto che il contributo dei pazienti alla scoperta, sviluppo e valutazione dei farmaci arricchisca la qualità delle evidenze e delle opinioni disponibili.[1]
I codici esistenti di comportamento per il coinvolgimento dei pazienti con le varie parti in causa non coprono in modo esaustivo l’intera portata della ricerca e dello sviluppo (research and development, R&D). I documenti di orientamento di EUPATI hanno lo scopo di supportare l’integrazione del coinvolgimento dei pazienti nell’intero processo di ricerca e sviluppo dei farmaci.
Questi documenti di orientamento non hanno come fine di essere prescrittivi e non daranno suggerimenti dettagliati passo passo.
EUPATI ha sviluppato questi documenti di orientamento per tutte le parti in causa che hanno come obiettivo quello di interagire coi pazienti nella ricerca e nello sviluppo dei farmaci (R&D). Gli utenti possono deviare da tale orientamento secondo specifiche circostanze, normative nazionali o le necessità esclusive di ciascuna interazione. Quest’orientamento deve essere adattato a requisiti individuali utilizzando il miglior giudizio professionale.
Vi sono quattro diversi documenti di orientamento che si occupano del coinvolgimento dei pazienti nei seguenti campi:
- R&D dei farmaci condotto dall’industria farmaceutica
- Comitati etici
- Autorità di regolamentazione
- Valutazione delle tecnologie sanitarie (HTA)
Ciascun orientamento suggerisce aree in cui al momento vi sono opportunità per il coinvolgimento dei pazienti. Quest’orientamento deve essere periodicamente rivisto e revisionato per rifletterne l’evoluzione.
Quest’orientamento si occupa del coinvolgimento dei pazienti nel campo normativo e fa riferimento al “Quadro di riferimento per l’interazione tra l’Agenzia europea per i medicinali e pazienti e consumatori e le loro organizzazioni”.
I seguenti valori sono riconosciuti nell’orientamento e hanno lavorato in direzione dell’adozione delle pratiche di lavoro suggerite (sezione 7). I valori sono i seguenti:
| Importanza | I pazienti hanno conoscenze, prospettive ed esperienze che sono uniche e contribuiscono in modo significativo ad aspetti fondamentali delle attività di regolamentazione. |
| Imparzialità | I pazienti hanno gli stessi diritti nel contribuire alle attività di regolamentazione dei farmaci delle altre parti in causa e hanno accesso a conoscenze ed esperienze che permettono un coinvolgimento efficace. |
| Equità | Il coinvolgimento dei pazienti nelle attività di regolamentazione contribuisce all’equità tramite la ricerca della comprensione dei diversi bisogni dei pazienti con particolari problemi sanitari, rispetto ai severi requisiti della normative e delle linee guida relative alla regolamentazione. |
| Legittimità | Il coinvolgimento dei pazienti facilita coloro che sono affetti da decisioni normative al fine di partecipare alle attività di regolamentazione, contribuendo alla trasparenza, responsabilità e credibilità del processo decisionale. |
| Costruzione di competenze | I processi di coinvolgimento dei pazienti affrontano gli ostacoli al coinvolgimento dei pazienti nelle attività di regolamentazione e costruiscono capacità di collaborazione tra pazienti ed enti di regolamentazione. |
Tutto l’orientamento successivamente sviluppato deve essere allineato alla normativa nazionale esistente che si occupa delle interazioni come dichiarato nei quattro documenti di orientamento di EUPATI.
Dichiarazione di limitazione di responsabilità
EUPATI ha sviluppato questo documento di orientamento per tutte le parti in causa che hanno come obiettivo quello di interagire coi pazienti nella ricerca e nello sviluppo dei farmaci (R&D) in tutto il ciclo di vita di R&D dei farmaci.
Questi documenti di orientamento non hanno come fine di essere prescrittivi e non daranno suggerimenti dettagliati passo passo. Tale orientamento deve essere utilizzato secondo specifiche circostanze, normative nazionali o le necessità esclusive di ciascuna interazione. Quest’orientamento deve essere adattato a requisiti individuali utilizzando il miglior giudizio professionale.
Dove questo orientamento offre consigli su problemi legali, non è offerto come definitiva interpretazione legale e non è sostitutivo di una consulenza legale formale. Se è necessaria una consulenza formale, le parti interessate devono consultare il rispettivo dipartimento di affari legali se disponibile o ricercare consulenza legale da parte di fonti competenti.
EUPATI non sarà in alcun caso responsabile di eventuali risultati di qualsiasi natura originati dall’uso di tale orientamento.
Il progetto EUPATI ha ricevuto supporto dall’Impresa congiunta dell’Iniziativa in materia di medicinali innovativi secondo l’accordo n° 115334, le cui risorse sono costituite da contributi finanziari da parte del Settimo programma quadro dell’Unione Europea (FP7/2007-2013) e delle aziende della Federazione europea delle associazioni dell’industria farmaceutica (European Federation of Pharmaceutical Industry Associations, EFPIA).
Scopo
Quest’orientamento europeo si occupa delle interazioni tra i pazienti e gli enti di regolamentazione dei farmaci in relazione ai farmaci per uso umano. I “pazienti” possono essere pazienti singoli o i loro prestatori di cure, o rappresentanti provenienti da organizzazione dei pazienti con esperienze importanti (sezione 4). Gli enti di regolamentazione includono l’Agenzia europea per i medicinali (EMA) e le Autorità nazionali competenti (National Competent Authorities, NCA). Le organizzazioni dei pazienti sono organizzazioni no profit che hanno interesse alla cura dei pazienti, e dove i pazienti rappresentano una maggioranza dei membri presenti negli organismi di governo.
L’orientamento si focalizza sul coinvolgimento ed esclude la raccolta scientifica di prospettive dei pazienti (vale a dire, una ricerca sistematica dal punto vista quantitativo e qualitativo sull’impatto psicosociale delle malattie e dei trattamenti). La Figura 1 indica le fasi in cui i pazienti possono essere attualmente coinvolti nel corso di tutto il ciclo di vita di R&D dei farmaci; tuttavia, ciò non deve significare un limite al coinvolgimento, e tali opportunità possono modificarsi e accrescersi nel corso del tempo.
Definizione di "paziente"
Il termine "paziente" viene spesso utilizzato come termine generale, impreciso, che non riflette i diversi tipi di input ed esperienze richieste da pazienti, difensori dei pazienti e organizzazioni dei pazienti in diversi processi collaborativi.
Al fine di chiarire la terminologia per ruoli potenziali dell'interazione dei pazienti presentati in questi e altri documenti di orientamento di EUPATI, utilizziamo il termine "paziente" per coprire le seguenti definizioni:
- I "pazienti singoli" sono persone con esperienza personale di vita con una malattia. Possono o non possono possedere conoscenze tecniche riguardo a R&D o ai processi di regolamentazione, ma il loro ruolo principale è di contribuire con l'esperienza soggettiva personale della malattia e del trattamento.
- I "prestatori di cure" sono persone che assistono pazienti singoli, come familiari nonché aiutanti volontari o pagati.
- I "difensori dei pazienti " sono persone che hanno osservazioni ed esperienza in relazione al supporto di una più grande popolazione di pazienti che vivono con una malattia specifica. Possono o non possono essere affiliati a un'organizzazione.
- I "rappresentanti di organizzazioni dei pazienti" sono persone autorizzate a rappresentare ed esprimere punti di vista collettivi di un'organizzazione di pazienti su un problema o su un'area patologica specifica.
- I "pazienti esperti", oltre ad avere competenze su una specifica malattia, hanno le conoscenze tecniche in R&D e/o affari normativi tramite formazione o esperienze, ad esempio membri di EUPATI che sono stati formati da EUPATI sull'intero spettro di R&D dei farmaci.
Possono esservi riserve sul coinvolgimento di singoli pazienti in attività di collaborazione con portatori d'interesse per il motivo che il loro input è soggettivo e aperto a critica. Tuttavia, EUPATI, in linea con gli enti di regolamentazione, istilla il valore dell'equità non escludendo il coinvolgimento di singoli. È necessario che venga lasciato alla discrezione dell'organizzazione/delle organizzazioni che avviano l'interazione la scelta della rappresentanza più adeguata di pazienti nei termini di quale tipo di paziente e per quale attività (vedere sezione 7). Dove un paziente singolo sarà impegnato, si suggerisce che le relative organizzazioni dei pazienti, dove ne esiste una, siano informate e/o consultate al fine di fornire supporto e/o consiglio.
Il tipo di input e mandato della persona interessata deve essere concordato in eventuali processi collaborativi precedenti il coinvolgimento.
Base logica per l'orientamento
La portata del coinvolgimento dei pazienti nei problemi normativi varia in modo considerevole tra paesi e regioni in Europa.
L'EMA ha interagito con le sue parti in causa dalla sua creazione nel 1995. Tali relazioni tra parti in causa si sono evolute nel corso del tempo e il tipo e l'entità dell'interazione varia secondo il gruppo di parti in causa interessato e il tipo di attività di EMA. Il Comitato di gestione dell'EMA e certi comitati scientifici comprendono come membri pazienti e consumatori.
Il beneficio del coinvolgimento delle parti in causa sperimentato dall'EMA ha avuto come esito l'implementazione di un quadro per il coinvolgimento dei pazienti anche a livello nazionale da parte di diversi enti nazionali di regolamentazione. La maggioranza degli enti di regolamentazione nazionali si basa sulle esperienze dell'EMA. Il coinvolgimento dei pazienti con l'EMA viene definito dalla normativa europea [2]. L'EMA, il suo Comitato di gestione e i suoi vari comitati sono responsabili per lo sviluppo delle relazioni tra l'EMA e i suoi portatori d'interessi.
La normativa europea definisce i seguenti aspetti:
- L'interazione diretta tra l'EMA e le organizzazioni di pazienti e di consumatori tramite il gruppo di lavoro di pazienti e consumatori dell'EMA (Patients' and Consumers' Working Party, PCWP).
- Il quadro di riferimento che permette di fornire informazioni chiare e utili a tali organizzazioni.
- Forme specifiche d'interazione, ad es. partecipazione dei pazienti in qualità dei membri nel Comitato di gestione dell'EMA, il Comitato per i prodotti medicinali orfani (Committee for Orphan Medicinal products, COMP), il Comitato pediatrico (Paediatric Committee, PDCO), il Comitato per le terapie avanzate (Committee for Advanced Therapies, CAT), in procedure di consulenza scientifica/assistenza in relazione al protocollo con il Gruppo di lavoro di consulenza scientifica (Scientific Advice Working Party, SAWP) e il Comitato di valutazione del rischi per la farmacovigilanza (Pharmacovigilance and Risk Assessment Committee, PRAC).
- Inoltre, l'EMA ha introdotto dei metodi per la raccolta delle opinioni tramite consultazione diretta.
L'esperienza acquisita ad oggi dimostra che la partecipazione dei pazienti nelle attività di EMA ha avuto come esito una maggiore trasparenza e fiducia nei processi normativi e un maggiore rispetto reciproco tra enti di regolamentazione e la comunità di pazienti e consumatori. L'esperienza conferma l'importanza per l'EMA nel continuare a supportare e facilitare il contributo dei pazienti al suo lavoro.
Simili disposizioni normative possono mancare a livello nazionale. In assenza di disposizioni normative, le Autorità competenti nazionali stanno sviluppando un loro quadro di riferimento sulla base dell'esperienza dell'EMA o stanno sviluppandone uno proprio. Gli elementi chiave da considerare in tale ambito sono i seguenti:
- Definizione del ruolo dei pazienti nell'interazione
- Inclusione di proposte sul coinvolgimento dei pazienti in specifici processi decisionali
- Sviluppo di un programma di formazione
- Presa in considerazione di un concetto per il compenso di esperti, applicato a tutte le parti in causa
- Valutazione continua dell'interazione per ulteriori miglioramenti e collaborazione tra agenzie con i pazienti al fine di stabilire e standardizzare metodi e pratiche.
Qualsiasi quadro di riferimento necessita di essere rivisto su base regolare.
Obiettivi del coinvolgimento dei pazienti nella regolamentazione dei farmaci
L'ottimizzazione delle interazioni coi pazienti e la focalizzazione su aree dove può essere previsto un mutuo beneficio sono due principi sottostanti da considerare quando si applica un quadro di riferimento.
Lo scopo deve essere l'ulteriore sviluppo di trasparenza e fiducia con le comunità di pazienti tramite il loro impegno attivo (partecipazione- consultazione-informazione) Al fine di ottenere tale obiettivo, devono essere soddisfatti obiettivi specifici, tra cui i seguenti:
- Supportare l'ente di regolamentazione al fine di accedere a esperienze reali di malattie e della loro gestione e di ottenere informazioni sull'attuale uso di farmaci. Ciò contribuirà a comprendere il valore, come percepito dai pazienti, delle evidenze scientifiche fornite durante il processo di valutazione ai fini del processo decisionale relativo al rapporto benefici/rischi.
- Garantire che i pazienti e le loro organizzazioni di rappresentanza siano ascoltati, consultati e coinvolti nello sviluppo di politiche e di piani.
- Migliorare la comprensione da parte delle organizzazioni di pazienti del mandato e del ruolo dell'ente di regolamentazione entro il contesto dello sviluppo, valutazione, autorizzazione, monitoraggio e la comunicazione d'informazioni relative ai farmaci.
- Ottimizzare gli strumenti di comunicazione (sui contenuti e sulla prestazione) al fine di facilitare e incoraggiare la cascata d'informazioni verso i componenti delle organizzazioni di pazienti (vale a dire raggiungere i singoli pazienti) con lo scopo di supportare il loro ruolo nell'uso sicuro e razionale dei farmaci.
- Facilitare la partecipazione di pazienti alla valutazione benefici/rischi e attività correlate, al fine di registrare i valori e le preferenze dei pazienti e di ottenere informazioni sull'uso attuale di farmaci e sul loro ambiente terapeutico, durante tutto il ciclo di vita dello sviluppo di farmaci, a partire dallo sviluppo iniziale e nel corso di tutta la valutazione e la sorveglianza a seguito dell'immissione in commercio.
Il raggiungimento di tali obiettivi necessita di una stretta collaborazione tra enti di regolamentazione, ministeri nazionali della salute e altre importanti parti in causa, nonché una partecipazione attiva e una buona interazione coi pazienti, i professionisti sanitari e le loro organizzazioni di rappresentanza.
Pratiche di lavoro suggerite (adattato dal quadro di riferimento dell'EMA sulle interazioni)
Sulla base dell'esperienza dell'EMA a livello europeo, i pazienti possono partecipare alle attività degli enti di regolamentazione come:
- membri (e alternati) di alcuni dei comitati (scientifici) o gruppi di lavoro degli enti di regolamentazione e, nel caso dell'EMA, del Comitato di gestione dell'EMA (nominato formalmente dalle istituzioni dell'UE);
- esperti singoli;
- rappresentanti di un'organizzazione specifica, che vengano consultati e partecipino in dibattiti per esprimere i punti di vista dell'organizzazione su un problema specifico;
- occasionalmente osservatori in certi aspetti del lavoro dell'EMA e degli enti di regolamentazione.
Gli enti di regolamentazione devono stabilire criteri d'idoneità.
Quando i pazienti partecipano alle attività di regolamentazione come individui e non come rappresentanti delle loro organizzazioni, devono dichiarare eventuali interessi e attenersi al codice di condotta dell'ente di regolamentazione come ogni altro esperto. Inoltre, le organizzazioni coinvolte con gli enti di regolamentazione devono essere completamente trasparenti riguardo alle loro attività e alle loro fonti di finanziamento.
Al fine di ottenere gli obiettivi individuati dalla sezione 4, devono essere presi in considerazione come cruciali i seguenti sei elementi:
- Una rete di organizzazioni di pazienti (potenzialmente in collaborazione con altri enti di regolamentazione)La rete di organizzazioni di pazienti consente all'ente di regolamentazione di costruire interazioni coerenti e mirate con un ampio gruppo di organizzazioni con un'ampia gamma di esperienze e di interessi. Devono applicarsi dei criteri di selezione. Tali criteri devono garantire che gli enti di regolamentazione stabiliscano dei contatti con le organizzazioni più adatte che rappresentino i pazienti in maniera trasparente. In una rete, i criteri devono essere armonizzati.
- Un forum di scambio con organizzazioni di pazienti stabilito entro l'ente di regolamentazioneQuesta è una piattaforma per il dialogo e per lo scambio con organizzazioni di pazienti su problemi pertinenti che riguardano i farmaci per uso umano e nel caso dispositivi medici, attraverso di essa l'ente di regolamentazione informerà e otterrà feedback e contributi da parte di pazienti in varie iniziative dell'ente di regolamentazione. Include una rappresentazione equilibrata dei diversi tipi di pazienti nonché organizzazioni che rappresentano popolazioni speciali e vulnerabili non ben rappresentate nello sviluppo dei farmaci come persone anziane e donne. È necessario disporre di un forum per individuare ulteriormente lacune e priorità nelle interazioni a livello globale.
- Un pool di pazienti singoli che agiscono come esperti nella loro malattia e nel suo trattamento al fine di facilitare il coinvolgimento dei pazienti nella valutazione e nell'informazione dei pazienti.La creazione del pool di esperti consentirà agli enti di regolamentazione di identificare rapidamente ed efficientemente pazienti che possono essere coinvolti in attività relative al prodotto, rivedere informazioni relative al prodotto e materiale di comunicazione.
- Interazione in particolare nel campo della comunicazione Ciò fornirà un contributo valido per il supporto di strutture esistenti per la divulgazione di informazioni al pubblico. Inoltre, la collaborazione in quest'area promuoverà l'offerta di informazioni valide e aggiornate ai pazienti sui benefici e sui rischi dei farmaci e contribuirà alla preparazione e alla divulgazione di messaggi chiari volti a raggiungere il pubblico sull'uso sicuro e razionale di farmaci. Qualsiasi materiale informativo per i pazienti deve essere rivisto da rappresentanti dei pazienti al fine di migliorare la leggibilità e l'adeguatezza del linguaggio e dei contenuti.
- Un programma di azioni per la costruzione di competenze, che si focalizzi sulla formazione e sull'accrescimento della consapevolezza circa il sistema normativo Affinché il loro contributo sia significativo, i pazienti devono avere una comprensione del mandato dell'ente di regolamentazione nonché del ruolo atteso dei pazienti nel processo di valutazione. Deve essere disponibile un programma di formazione. Alcune organizzazioni di pazienti o altri progetti collaborativi hanno sviluppato il proprio materiale per la formazione al fine di rafforzare i pazienti per avere un ruolo riconosciuto di difesa.
- Supporto finanziario Ai pazienti che contribuiscono alle attività di regolamentazione deve essere fornito supporto finanziario. Ciò rappresenterà un riconoscimento del lavoro da loro svolto e allo stesso tempo promuoverà la loro indipendenza. I pazienti devono essere riconosciuti come esperti e trattati con gli stessi standard di qualsiasi altro esperto, anche riguardo al compenso. Talvolta, i pazienti possono aver bisogno di assistenza supplementare per garantire che siano in grado di partecipare.
Definizione delle interazioni
Prima di ciascuna interazione, è necessario concordare mutualmente sui seguenti aspetti (ove applicabile):
- L'obiettivo dell'attività che coinvolge i pazienti e/o le aree di interesse comune per stabilire un'interazione strutturata concordata, che fornisca a tutte le parti la tutela necessaria riguardo all'indipendenza, alla riservatezza e alle aspettative.
- Il tipo di input e mandato della persona interessata.
- Gli strumenti e i metodi d'interazione, ad es. frequenza degli incontri, regole di base, risoluzione di conflitti, compenso, valutazione.
- Il metodo d'interazione (incontri, discussioni per telefono ecc.) deve essere discusso e concordato, essendo la priorità principale la convenienza dei pazienti/delle organizzazioni di pazienti. Se l'interazione richiede incontri di persona o lo sviluppo e l'offerta di eventi, questi ultimi devono seguire codici esistenti di condotta, normative locali, in termini di adeguatezza del locale/del luogo e del livello di ospitalità fornito.
- Quando vengono organizzati degli eventi, è necessario prendere in considerazione la partecipazione attesa del pubblico, con misure appropriate al fine di consentire l'accessibilità, l'assistenza negli spostamenti e l'ammissione agli eventi.
- Paziente e organizzazione di pazienti partner desiderati per la promozione di relazioni di collaborazione a lungo termine, con indipendenza garantita.
- Il profilo del tipo di paziente/i o di rappresentante/i di pazienti da coinvolgere e il loro numero.
- In che modo saranno utilizzati gli output dell'attività.
- In che modo e quando il paziente/i pazienti interessato/i sarà/saranno informati degli outcome.
- Termini e condizioni contrattuali inclusi il consenso e la riservatezza nonché l'accordo sull'interazione stessa (tipo di incontro, frequenza, compenso).
- Altri elementi secondo l'attività specifica.
Individuazione/interazione del paziente
Vi sono molti modi per identificare i pazienti da coinvolgere in un'interazione. I percorsi principali si svolgono tramite:
- organizzazioni di pazienti esistenti
- EUPATI o progetto simile
- opportunità di pubblicità per la partecipazione dei pazienti
- chiamata aperta
- relazioni esistenti con operatori sanitari, ospedali e ricercatori e altre agenzie
- richieste non sollecitate precedentemente fatte da parti interessate
- comitati/gruppi consultivi esistenti (ad es, Gruppo di lavoro di pazienti e consumatori presso l'EMA, Think Tank di EFPIA)
- agenzie terze.
Criteri d'idoneità
Organizzazioni di pazienti
Al fine di accrescere la trasparenza del coinvolgimento dei pazienti le agenzie e le organizzazioni dei pazienti devono pianificare di divulgare pubblicamente le proprie attività di collaborazione su base annuale. Nomi individuali di pazienti possono essere divulgati quando la persona fa parte di un comitato consultivo generico ma in altri casi i nomi non devono essere divulgati.
Le organizzazioni di pazienti si devono impegnare a svolgere una parte attiva nell'interazione con un ente di regolamentazione.
Le organizzazioni saranno stabilite in uno Stato membro dell'Unione Europea (UE) o dello Spazio economico europeo (SEE) e dovranno soddisfare i seguenti criteri:
| Legittimità: | l'organizzazione dovrà disporre di statuti registrati in uno degli Stati membri dell'UE/SEE. Se si tratta di un'organizzazione internazionale non registrata in uno Stato membro dell'UE/SEE, devono essere fornite informazioni aggiuntive che dimostrano il focus e le attività nell'UE. |
| Obiettivi/missioni: | l'organizzazione o il paziente esperto singolo deve definire in modo chiaro la sua/i suoi missione/obiettivi e deve concordare di pubblicarla/pubblicarli sul sito web dell'ente di regolamentazione. |
| Attività: | l'organizzazione avrà, tra le sue attività, un interesse specifico nei prodotti medicinali (e nel caso nei dispositivi medici), cosa che deve essere documentata (ad es. tramite un rapporto pubblicato sul sito web dell'organizzazione o delle persone individuali). |
| Rappresentatività: | l'organizzazione deve essere rappresentativa dei pazienti in tutta l'UE/tutto il SEE o al relativo livello nazionale. Si ritiene che le organizzazioni già registrate a livello di comunità, ad es, nel Forum della salute dell'UE, il Consiglio d'Europa, rappresentino adeguatamente i pazienti o siano adeguate per il coinvolgimento nelle attività di regolamentazione dei farmaci.
In caso di mancanza di associazioni europee per una malattia specifica o una specifica area di trattamento, può essere preso in considerazione il coinvolgimento di organizzazioni nazionali, sebbene sia data la preferenza ad associazioni di livello europeo. Ai fini dell'idoneità, possono essere prese in considerazione organizzazioni internazionali purché abbiano un focus e una rappresentativa europea, inclusi ufficio/uffici con sede in UE/SEE. |
| Struttura: | l'organizzazione deve disporre di organismi di governo che siano eletti dai propri membri, i quali saranno pazienti, i loro prestatori di cure o i loro rappresentanti eletti. |
| Responsabilità e modalità di consultazione: | dichiarazioni e opinioni dell'organizzazione devono riflettere i punti di vista e le opinioni dei loro membri e devono essere introdotte procedure adeguate di consultazione con quei membri. In particolare, le organizzazioni devono garantire che vi sia un adeguato flusso d'informazioni per permettere il dialogo in entrambe le direzioni: da e verso i suoi membri. |
| Trasparenza: | l'organizzazione divulgherà agli enti di regolamentazione le proprie fonti di finanziamento sia pubbliche che private fornendo il nome degli organismi e il loro contributo finanziario individuale, sia in termini assoluti che in termini di percentuale globale di budget organizzativo. Eventuali relazioni con sponsor aziendali devono essere chiare e trasparenti. Tali informazioni saranno comunicate all'Agenzia su base annuale.
Nel caso di organizzazioni di riferimento, l'elenco delle associazioni che vi aderiscono deve essere reso disponibile all'Agenzia. L'organizzazione pubblicherà sul sito web dell'organizzazione gli statuti registrati, insieme alle informazioni finanziarie comprese le sue fonti di finanziamento sia pubblico che privato, e le informazioni riguardanti le loro attività. L'organizzazione seguirà un codice di condotta/politiche che regolano la sua relazione con gli sponsor e la sua indipendenza da esso. L'ente di regolamentazione valuterà le informazioni di natura finanziaria secondo un regolamento prefissato trasparente. |
Compenso
È necessario riconoscere che in molte situazioni i pazienti coinvolti nelle attività lo fanno volontariamente come singoli ma anche come membri di un'organizzazione. È necessario quindi considerare:
- un compenso per il tempo totale investito più le spese;
- eventuali compensi offerti devono essere giusti e adeguati per il tipo di impegno. Idealmente i costi di viaggio dovrebbero essere pagati direttamente dal partner organizzatore, piuttosto che essere rimborsati;
- la copertura dei costi incorsi dalle organizzazioni dei pazienti nell'identificazione o nel supporto dei pazienti per il coinvolgimento in attività (ad es. gruppi di pari di supporto, formazione e preparazione);
- aiuto nell'organizzazione della logistica della partecipazione dei pazienti, inclusi spostamenti e/o sistemazione.
Il compenso comprende anche benefici indiretti (come la fornitura di servizi gratuiti da parte di un'organizzazione di pazienti) o qualsiasi altro beneficio non finanziario fornito al paziente/all'organizzazione dei pazienti (come sessioni di formazione, servizi di agenzia, la realizzazione di siti web).
Accordo scritto
Un accordo scritto deve definire chiaramente almeno i seguenti aspetti: una descrizione dell'attività e dei suoi obiettivi, la natura dell'interazione durante l'attività, il consenso (se pertinente), la pubblicazione, la riservatezza, il compenso, la riservatezza dei dati, la compliance, la dichiarazione dei diritti di interesse, le linee guida. L'interazione può procedere solamente sulla base di un accordo scritto che come minimo dichiari precisamente gli elementi di base della collaborazione (ad es. le regole riguardanti il coinvolgimento, la compliance, la proprietà intellettuale, i compensi finanziari). È necessario porre attenzione in modo che gli accordi scritti siano chiari e non limitino la condivisione appropriata delle conoscenze.
Implementazione e monitoraggio
Un quadro di riferimento sul coinvolgimento dei pazienti può essere introdotto passo passo e/o seguire una fase pilota se appropriato. Dopo un'implementazione completa, quando i pazienti sono coinvolti sia in problemi specifici che generali relativi al prodotto e vi è un determinato pool di organizzazioni e di pazienti come individui esperti, nonché per le interazioni, è necessario preparare un rapporto pubblico annuale sulle interazioni che comprenda i seguenti aspetti:
- un'analisi degli indicatori (da definire secondo il tipo di interazione) che valuti l'utilità delle interazioni;
- feedback ricevuto dai pazienti e dalle loro organizzazioni rappresentative mediante indagini mirate;
- feedback ricevuto da parte dell'ente di regolamentazione stesso;
- un sommario delle attività in cui sono stati coinvolti le organizzazioni e i pazienti come esperti individuali;
- si raccomanda di proporre un suggerimento relativo alle modalità di proseguimento dell'attività, inclusa una strategia per interazione future di pazienti.
Appendice 1 - Roadmap sul coinvolgimento dei pazienti nei processi normativi - esempio EMA
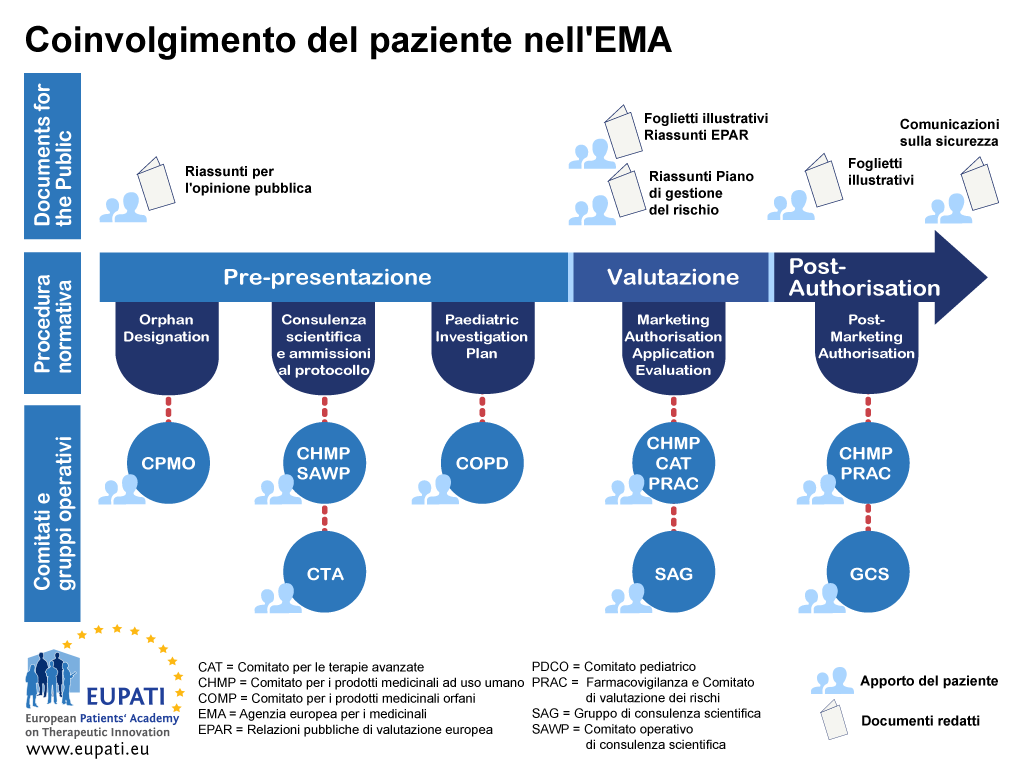
Appendice 2 – Codici di comportamento rivisti
Una serie di codici riconosciuti potrebbe fornire un fondamento importante per questo documento di orientamento.
- Il protocollo del Comitato consultivo della Comunità europea (ECAB), gruppo scientifico di lavoro presso il Gruppo europeo di trattamento dell'AIDS (EATG), fondato nel 1997 (descrizione di ECAB e delle sue procedure operative).
- Mandato, obiettivi e regole di procedura per il Gruppo di lavoro con organizzazioni dei pazienti e dei consumatori (PCWP) dei Comitati scientifici umani dell'Agenzia europea dei medicinali (30 maggio 2013)
- Verbale dell'incontro con tutte le parti idonee del Gruppo di lavoro con organizzazioni dei pazienti e dei consumatori (PCWP) dei Comitati scientifici umani dell'Agenzia europea dei medicinali (31 gennaio 2014)
- Documento di riflessione dell'EMA del 10 dicembre 2009 sull'ulteriore coinvolgimento di pazienti e consumatori nelle attività dell'agenzia
- Opuscolo dell'EMA sul lavoro coi pazienti e i consumatori (aggiornato 22/04/2015)
- Quadro di riferimento dell'EMA sulle interazioni (rivisto 16 ottobre 2014)
- Raccomandazioni da parte dell'incontro di ECAB tenutosi a Bergen, Norvegia, 1997ECAB di EATG, “The impatient Patient - From Anger to Activism” ("Il paziente impaziente -Dalla rabbia all'attivismo")Una revisione sistematica della storia, dei modelli di lavoro, dell'importanza e delle prospettive del Comitato consultivo della comunità europea
- Programma relativo ai rappresentanti dei pazienti della FDA
- Sviluppo dei farmaci centrato sul paziente della FDA; la Voce del paziente: Serie di report dell'Iniziativa dello sviluppo dei farmaci centrato sui pazienti della FDA
- Sviluppo dei farmaci centrato sul paziente della FDA: Miglioramento della valutazione dei rischi e dei benefici nel processo decisionale normativo
- Dichiarazione di Helsinki dell'Associazione medica mondiale (AMM) - Principi etici per la ricerca medica su soggetti umani Retrieved 13 July, 2021, from https://www.wma.net/policies-post/wma-declaration-of-helsinki-ethical-principles-for-medical-research-involving-human-subjects/
Riferimenti bibliografici
- Adapted from the EMA framework. European Medicines Agency (2014) EMA/637573/2014. http://www.ema.europa.eu/docs/en_GB/document_library/Other/2009/12/WC500018013.pdf. Last Accessed 21.11.2016.
- Regulation (EC) No 726/2004 http://ec.europa.eu/health/files/eudralex/vol-1/reg_2004_726/reg_2004_726_en.pdf. Last Accessed 21.11.2016.
*I consumatori sono riconosciuti come portatori di interesse all’interno del dibattito sanitario. L’obiettivo di Eupati si concentra sui pazienti piuttosto che sui consumatori come si evince dal materiale educazionale e dai documenti di orientamento.