Niet-klinische ontwikkeling: Basisprincipes
Inleiding
De niet-klinische (of preklinische) ontwikkelingsfase richt zich voornamelijk op het vaststellen van welke kandidaat-therapie de grootste kans op succes heeft, beoordeelt de veiligheid ervan en legt vóór overgang naar de klinische ontwikkelingsfase solide wetenschappelijke fundamenten.
Tijdens de niet-klinische ontwikkelingsfase moet de kandidaat-stof ook voldoen aan niet-medische doelstellingen, zoals definiëring van intellectuele-eigendomsrechten en het fabriceren van voldoende van het geneesmiddel voor klinisch onderzoek. De niet-klinische ontwikkeling van een geneesmiddel is complex en onderworpen aan regelgeving.
Basisprincipes, belangrijke definities en concepten
‘Niet-klinisch’ of ‘preklinisch’?
De termen ‘niet-klinisch’ en ‘preklinisch’ worden vaak door elkaar gebruikt.
Hoewel van essentieel belang bij de preklinische stappen van de ontwikkeling, kunnen niet-klinische onderzoeken op elk moment tijdens de levenscyclus van het middel worden uitgevoerd. Het is belang de preklinische stappen zo vroeg mogelijk uit te voeren om verrassingen later in de ontwikkeleing te voorkomen.
Behalve identificatie van de farmacodynamiek (wat een geneesmiddel doet met het lichaam), farmacokinetiek (wat het lichaam doet met het geneesmiddel) en toxicologie van de kandidaat-verbinding vóór toediening aan mensen, worden de gegevens van niet-klinische onderzoeken gebruikt voor verfijning, consolidatie en toevoeging van informatie voor het updaten van het veiligheidsprofiel van het middel tijdens de preklinische fase, op het moment van registratie en gedurende de levenscyclus van het geneesmiddel.
In-silico-, in-vitro-en in-vivo- onderzoeken
De onderzoeken tijdens de niet-klinische ontwikkeling worden als volgt uitgevoerd:
- In silico: ‘uitgevoerd op een computer of via computersimulatie’, bijv. het toxicologieprofiel van een middel voorspellen aan de hand van zijn chemische structuur afkomstig van op gegevens gebaseerde benaderingen.
- In vitro (Latijn voor ‘in het glas’): uitvoeren van een procedure in een gecontroleerde omgeving buiten een levend organisme, bijv. het gebruik van kweken van hepatocyten (levercellen) voor metabolismeonderzoek.
- In vivo (Latijn voor ‘in de levende’): experimenten uitvoeren met een intact, levend organisme in tegenstelling tot weefsels of cellen, d.w.z. dieren, mensen of planten.
Wat zijn de belangrijkste aspecten van chemie, fabricage en controle (‘Chemistry, Manufacturing, Control’, CMC) tijdens de niet-klinische ontwikkeling?
Alle niet-klinische ontwikkelingsonderzoeken vereisen de fabricage van een geschikte werkzame stof:
- Voor niet-klinische onderzoeken zijn doorgaans kleine hoeveelheden nodig (milligrammen tot grammen); om grotere hoeveelheden te produceren voor klinische onderzoeken en later, na goedkeuring, voor de markt moet vervolgens een opschalingsproces worden ontwikkeld.
- Voor ‘Good Laboratory Practice’ (GLP)-onderzoeken zijn gekwalificeerde of ‘Good Manufacturing Practice’ (GMP)-batches van de werkzame stof nodig.
Enkele belangrijke CMC-stappen in de niet-klinische ontwikkelingsfase zijn onder meer:
- Bepaling van de dosis en toedieningsweg
- Gedetailleerde fysisch-chemische karakterisering
- Stabiliteitstesten & onzuiverheidsanalyse
- Ontwikkelings- en validatiemethoden om de werkzame stof te kwantificeren in lichaamsvloeistoffen als bloed, plasma en urine in onderzoeken naar activiteit en bijwerkingen
- Ontwikkeling van een prototype van wat in de kliniek zal worden gebruikt.
Het niet-klinische ontwikkelingsproces
Niet-klinische ontwikkelingsactiviteiten parallel aan de onderzoeksactiviteiten Deze moeten het geplande klinische ontwikkelingsprogramma ondersteunen door in te gaan op de hieronder uiteengezette doelstellingen en vragen.
Doelstellingen
Zodra er een kandidaat-stof is geïdentificeerd, moet de niet-klinische ontwikkeling beginnen de volgende vragen te beantwoorden; de antwoorden komen dan van specifieke beoordelingen/onderzoeken:
- Werkt het? → werkzaamheidsbeoordeling
- Hoe wordt het afgegeven en hoe zal het lichaam reageren? → profilering
- Is het veilig? → toxicologie/veiligheid
- Is de fabricage uitvoerbaar en controleerbaar?
Niet-klinische ontwikkelingsactiviteiten kunnen gedurende de gehele levenscyclus van het middel worden voortgezet. Maar hoe eerder deze vragen worden beantwoord, hoe gemakkelijker het profiel van de patiënt kan worden vastgesteld die er het meest baat bij zal hebben.
Projectmanagement
Het niet-klinische ontwikkelingsprogramma is complex en vereist een solide projectmanagement en communicatieve vaardigheden bij de aansturende multidisciplinaire teams. Het projectteam moet het beoogde klinische plan begrijpen om het niet-klinische plan en hieraan gerelateerde activiteiten te definiëren.
Het profiel biedt een kader om de niet-klinische ontwikkelingsstrategie uit te voeren, doelen, risico’s, verantwoordelijkheden en meetmethoden te definiëren en te beslissen om de procedure wel of niet voort te zetten (zogenaamde “Go of No-Go” beslissing). Profielimplementatie helpt om de focus van het project op de belangrijkste productcriteria te houden, en tijdig te beslissen om wel of niet verder te gaan en om het algehele projectrisico te verlagen (d.w.z. voortzetting van de ontwikkeling van een niet-bruikbaar middel).
Niet-klinische regelgevingsrichtlijnen
Bij de ontwikkeling van geneesmiddelen zijn veel partijen betrokken en elke organisatie of instelling gaat te werk volgens de eigen regels. Zo hebben bedrijven hun standaard operationele procedures (SOP). Naast de voorschriften van ‘Good Clinical Practice’ kunnen op de website van het Europees Geneesmiddelenbureau (EMA) richtlijnen worden geraadpleegd.
- Dit zijn algemene of meer specifieke richtlijnen betreffende wetenschappelijke en technische aspecten (bijv. specifiek voor vereiste toxicologische onderzoeken).
- Deze moeten voor elke nieuwe vergunningaanvraag strikt worden nageleefd; elke afwijking moet worden verantwoord.
Gegevens worden gepresenteerd volgens het formaat van het gemeenschappelijke technische document (‘Common Technical Document’, CTD) zoals gedefinieerd door de Internationale Conferentie voor harmonisatie van de technische voorschriften voor de registratie van geneesmiddelen voor menselijk gebruik (ICH). De afspraak om alle informatie over kwaliteit, veiligheid en werkzaamheid samen te voegen in dit gemeenschappelijke formaat (het CTD) heeft voor een revolutie gezorgd inzake het voorgeschreven beoordelingsproces en heeft geleid tot geharmoniseerde elektronische vergunningaanvragen die, op hun beurt, de uitvoering van goede beoordelingspraktijken mogelijk maken. Voor de industrie heeft de afspraak de noodzaak weggenomen om de informatie voor een vergunningaanvraag aan te passen aan de verschillende toezichthoudende instanties (de IHC verenigt de toezichthoudende instanties en farmaceutische industrie van Europa, Japan en de VS voor overleg over wetenschappelijke en technische aspecten van geneesmiddelregistratie).
-
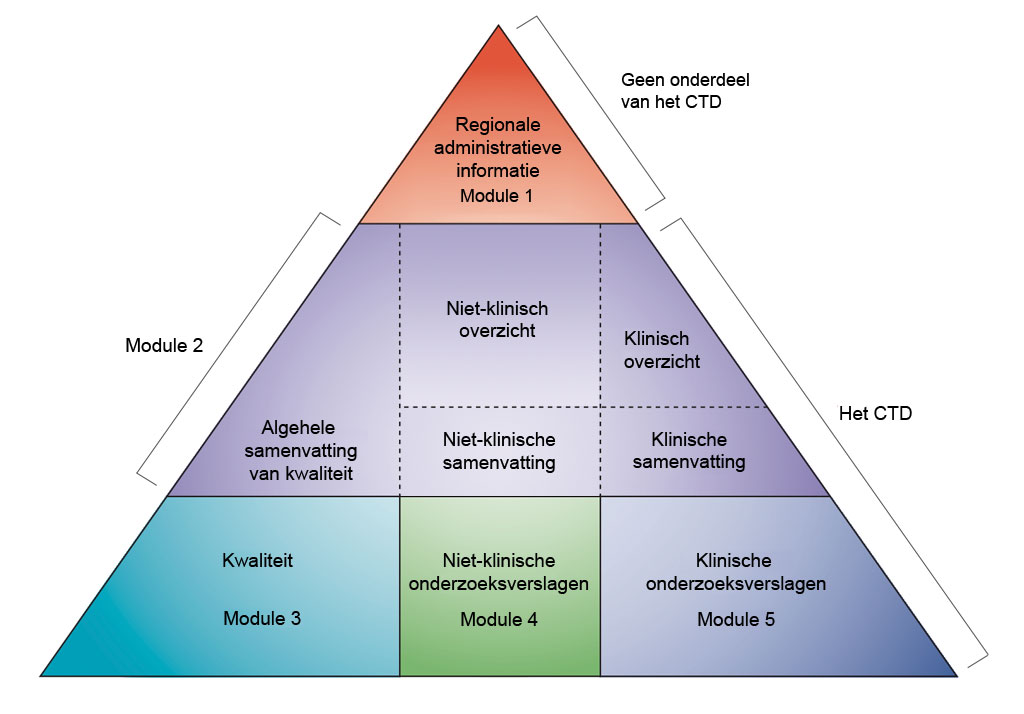
- Niet-klinische ontwikkeling in CTD-modules. Bewerkt naar ICH CTD (zie referentie 1)
Het CTD bestaat uit vijf modules (zie bovenstaande afbeelding). In juli 2003 werd het CTD het verplichte formaat voor nieuwe vergunningaanvragen in de EU en Japan, en het sterk aanbevolen formaat voor nieuwe toepassingen van geneesmiddelen (‘new drug applications’, NDA) ingediend bij de Amerikaanse Food and Drug Administration (FDA).
Samenvatting
De niet-klinische ontwikkelingsfase is cruciaal en moet anticiperen op potentiële problemen voordat een stof overgaat naar de klinische ontwikkelingsfase.
Voor opname van een kandidaat-verbinding in klinische onderzoeken is het volgende vereist:
- Niet-klinische veiligheidsbeoordeling verkregen onder omstandigheden volgens ‘Good Laboratory Practices’ (GLP).
- Fabricage uitgevoerd onder adequate kwaliteitscontrole.
- Gegevens en procedure gedocumenteerd volgens het CTD-formaat en fundamenten gelegd voor de klinische ontwikkelingsfase.
Er is sprake van een verhoogde tendens om geneesmiddelachtige eigenschappen in silico te ontwerpen, evenals om bio-informaticamethoden voor modellering en voorspelling toe te passen.
Richtlijnen voor niet-klinische ontwikkeling worden onderworpen aan continue harmonisatie tussen de belangrijkste toezichthoudende instanties (Europa, VS en Japan) met nadruk op de veiligheid en kwaliteit: ICH publiceert regelmatig gedetailleerde richtlijnen voor de farmaceutische industrie die vergelijkbaar zijn aan die welke gepubliceerd worden door Europese (EMA) en Amerikaanse (FDA) agentschappen.
Overige informatiebronnen
- European Medicines Agency (2007a). Guideline on strategies to identify and mitigate risks for first-in human trials with investigational medicinal products. London: European Medicines Agency. Retrieved 25 June, 2015, from http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002988.pdf
- European Medicines Agency (2007b). Guideline on requirements for first-in-man clinical trials for potential high-risk medicinal Retrieved 25 June, 2015, from http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002989.pdf
- European Medicines Agency (2009). ICH guideline M3(R2) on non-clinical safety studies for the conduct of human clinical trials and marketing authorisation for pharmaceuticals. London: European Medicines Agency. Retrieved 25 June, 2015, from http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002720.pdf
- European Medicines Agency (2011). Committee for medicinal products for human use (CHMP) ICH guidelines S6 (R1) – preclinical safety evaluation of biotechnology-derived pharmaceuticals. London: European Medicines Agency. Retrieved 25 June, 2015, from http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002828.pdf
[glossary_exclude]Referenties
- Image reproduced from ICH (2015). M4: The Common Technical Document. Retrieved 11 July, 2021, from https://www.ich.org/page/ctd
- European Medicines Agency (2009). ICH guideline M3(R2) on non-clinical safety studies for the conduct of human clinical trials and marketing authorisation for pharmaceuticals. London: European Medicines Agency. Retrieved 25 June, 2015, from http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002720.pdf[/glossary_exclude]
A2-2.01.1-1.2