Marktzulassung
Einleitung
Um legal im Europäischen Wirtschaftsraum (EWR) vermarktet werden zu können, braucht jedes Arzneimittel eine so genannte Genehmigung für das Inverkehrbringen, meist „Marktzulassung“ oder kurz „Zulassung“ genannt.1 Hinter diesem Genehmigungsprozess steht das Bestreben, den Bürgern des EWR zeitnah sichere, wirksame und qualitativ hochwertige Arzneimittel zur Verfügung zu stellen. Es muss darauf hingewiesen werden, dass die so genannten Medizinprodukte (Geräte, Instrumente und Ähnliches) dabei anderen Verfahren unterliegen als Arzneimittel.
Nationale Aufsichtsbehörden sind in den einzelnen Ländern dafür zuständig, die Anträge auf Marktzulassung zu überprüfen und die Zulassung zu erteilen, wenn die Zulassung des Arzneimittels im Rahmen eines nationalen Zulassungsverfahrens, einem dezentralisierten Zulassungsverfahren oder einem Verfahren der gegenseitigen Anerkennung angestrebt wird. Demgegenüber steht das zentralisierte Zulassungsverfahren, in dem die Anträge auf europaweite Marktzulassung von Arzneimitteln und deren wissenschaftliche Beurteilung von der Europäischen Arzneimittel-Agentur (EMA) in London koordiniert werden. Die Beurteilung wird von Sachverständigen der nationalen Aufsichtsbehörden durchgeführt. Wenn die Beurteilung positiv ausfällt, erteilt die Europäische Kommission die Marktzulassung.
Marktzulassungsverfahren
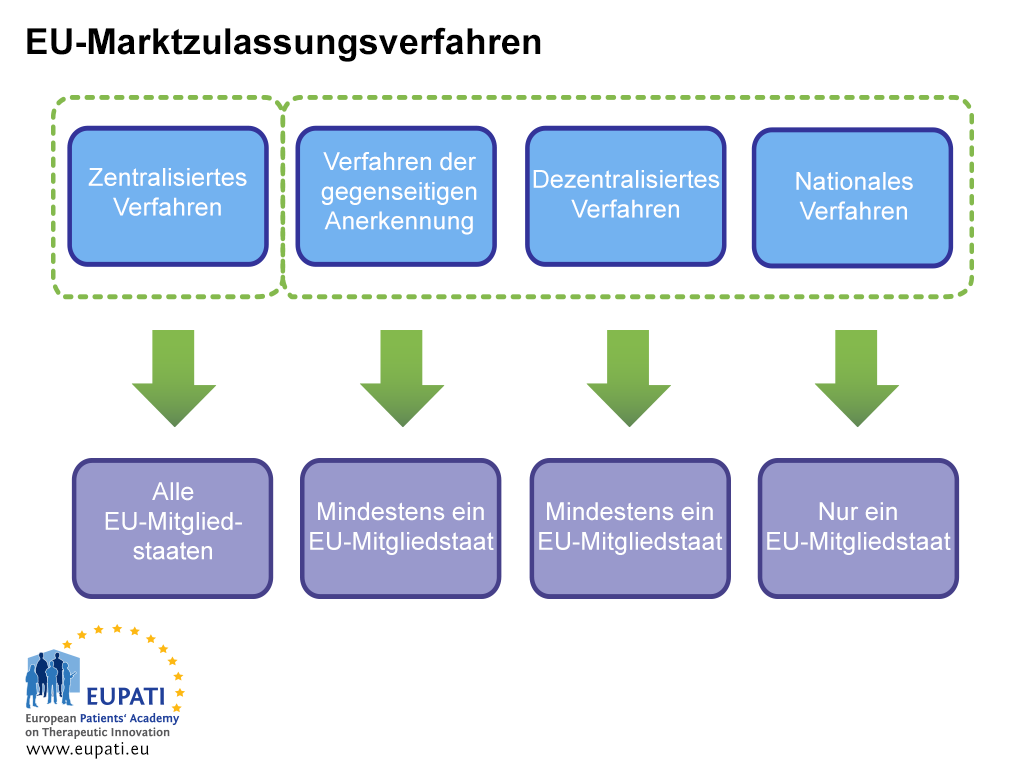
Eine Marktzulassung kann auf zwei Arten erwirkt werden: im zentralisierten Zulassungsverfahren oder in einem nicht zentralisierten Zulassungsverfahren. Zur zweiten Gruppe gehören das dezentralisierte Verfahren, das Verfahren der gegenseitigen Anerkennung und das nationale Verfahren. In jedem System gibt es eigene rechtliche Vorschriften und Verantwortlichkeiten für die zuständigen Behörden (EMA oder nationale Behörden) und Zulassungsinhaber.
Das zentralisierte Verfahren (CP)
Dies ist das Verfahren für die meisten neuen Arzneimittel. Das Unternehmen reicht einen einzigen Antrag bei der EMA ein. Das Komitee für Arzneimittel für die Anwendung am Menschen (CHMP) der EMA – bestehend aus je einem Mitglied für jeden der EU-Mitgliedstaaten und EWR-Länder sowie fünf wissenschaftlichen Sachverständigen – gibt eine Empfehlung an die Europäische Kommission aus, ob ein Arzneimittel genehmigt werden soll. Jedes Arzneimittel, das auf diese Weise genehmigt wurde, hat die Marktzulassung für alle Länder der EU und des EWR. Diese wissenschaftliche Beurteilung dauert höchstens 210 Arbeitstage, aber die Zählung kann auch angehalten werden (so genannte Aussetzung).
Dezentralisiertes Verfahren (DCP) und Verfahren der gegenseitigen Anerkennung (MRP)
In diesen Verfahren werden Marktzulassungen in mehreren Mitgliedstaaten gleichzeitig beantragt, wobei ein Land als Referenzmitgliedstaat (RMS) gilt. Wenn der Antrag erfolgreich ist, ist das Arzneimittel für die Vermarktung im RMS und den anderen beteiligten Ländern, den so genannten betroffenen Mitgliedstaaten (CMS), zugelassen. Diese Verfahren basieren auf dem Prinzip der Anerkennung der Einschätzung des RMS durch die CMS. Die meisten Generika werden mithilfe dieses Verfahrens zugelassen.
Nationales Verfahren
Unabhängige nationale Verfahren sind ausschließlich auf Arzneimittel beschränkt, die nur in einem Mitgliedstaat (MS) zugelassen und vermarktet werden sollen. Dieses Verfahren wird für neue Produkte nur noch selten angewendet.
Auswahl eines Marktzulassungsverfahrens
Normalerweise hat ein pharmazeutisches Unternehmen die freie Wahl, welches Verfahren es betreiben will. Diese unternehmerische Entscheidung fußt auf verschiedenen Vor- und Nachteilen der einzelnen Verfahren. Für folgende Produkte ist jedoch das CP verpflichtend:
- Produkte aus biotechnologischer Herstellung
- Arzneimittel für neuartige Therapien wie Gentherapien, Körperzellentherapeutika oder Arzneimittel aus Tissue Engineering (Gewebezüchtung)
- Arzneimittel für seltene Erkrankungen
- Arzneimittel für die Behandlung von HIV oder AIDS, Krebs, neurodegenerativen Erkrankungen, Autoimmunerkrankungen oder anderen Immunstörungen, Viruserkrankungen oder Diabetes
Das CP kann auch verwendet werden für Arzneimittel, die eine erhebliche therapeutische, wissenschaftliche oder technische Innovation darstellen oder im Interesse der öffentlichen Gesundheit sind, wie zum Beispiel Arzneimittel zum Abnehmen.
Wenn ein Unternehmen ein vorhandenes Arzneimittel, das bereits eine Marktzulassung hat, auch gegen eine andere Erkrankung („Indikation“) anwenden möchte, muss es im Allgemeinen eine vollständig neue Marktzulassung beantragen.
-
- An der Marktzulassung eines Arzneimittels sind verschiedene Parteien beteiligt, je nachdem, welches Verfahren der Sponsor für die Beantragung ausgewählt hat (oder befolgen muss).
Wissenschaftliche Beurteilung von Arzneimitteln
Unabhängig vom Verfahren muss für die Zulassung eines Arzneimittels ein „Dossier“ nach bestimmten Standards erstellt werden. Dieser standardisierte Antrag, das so genannte „Common Technical Document“ (CTD), ist ein international anerkanntes, harmonisiertes Format, das ein Antrag haben muss, wenn er bei den Zulassungsbehörden vorgelegt werden soll. Das CTD besteht eigentlich aus mehreren Dokumenten, mit denen der Antragsteller nachweisen muss, dass das Arzneimittel die erforderliche Qualität hat, sicher ist und wirksam ist.
Versagung der Zulassung
Eine Marktzulassung wird versagt (also nicht genehmigt), wenn
- das Arzneimittel ein ungünstiges Nutzen-Risiko-Verhältnis hat oder
- der Antragsteller keinen ausreichenden Nachweis der therapeutischen Wirksamkeit erbringt oder
- die qualitative und quantitative Zusammensetzung nicht den Angaben entspricht.
Gültigkeit der Zulassung und Nachkontrolle
Die Marktzulassung muss nach fünf Jahren auf der Basis einer Neubewertung des Nutzen-Risiko-Verhältnisses durch die Aufsichtsbehörde des genehmigenden Mitgliedstaates verlängert werden. Nach dieser Verlängerung ist die Zulassung unbegrenzt gültig, es sei denn, die zuständige Behörde entscheidet aus Gründen der Pharmakovigilanz auf einen weiteren fünfjährigen Verlängerungszeitraum.
Alle Inhaber von Marktzulassungen sind verpflichtet, auch nach der Zulassung weiterhin kontinuierlich Daten zur Unbedenklichkeit vorzulegen und andere Anforderungen einzuhalten. Sobald ein Arzneimittel auf dem Markt ist, muss es nämlich im Rahmen der so genannten Pharmakovigilanz weiter überwacht werden. Dabei wird nach Aspekten gesucht, die Auswirkungen auf das Sicherheitsprofil des Arzneimittels haben könnten, und es werden gegebenenfalls nach einer Untersuchung die notwendigen Maßnahmen ergriffen.
Nach der Marktzulassung können neue Informationen zur Wirksamkeit, Unbedenklichkeit oder Herstellung des Arzneimittels bekannt werden. In diesem Fall ist der Zulassungsinhaber dafür verantwortlich, den zuständigen Behörden einen Änderungsantrag zur gültigen Marktzulassung vorzulegen. Dieser geht an alle Mitgliedstaaten, die das Arzneimittel ursprünglich genehmigt haben.
Beteiligung von Patienten bei der Marktzulassung
Bisher ist die Beteiligung von Patienten an den Verfahren zur Marktzulassung sehr begrenzt. Einige zuständige Behörden denken aber darüber nach, wie Patienten stärker an Marktzulassungsverfahren beteiligt werden können – zum Beispiel durch Anhörung und/oder Vertretung von Patienten in der Commission on Human Medicines in der Behörde Medicines and Healthcare products Regulatory Agency (MHRA) im Vereinigten Königreich. Bereits beteiligt sind Patienten an der Nutzen/Risiko-Diskussion bei der EMA als Mitglieder des Ausschusses für Risikobewertung im Bereich der Pharmakovigilanz (PRAC), durch wissenschaftliche Beratung sowie Unterstützung bei der Erstellung von Prüfplänen und bei Sitzungen von wissenschaftlichen Beratungsgruppen. Es gibt auch eine Pilotstudie zur Beteiligung von Patienten im CHMP an den Anhörungen vor den Entscheidungen.
Patientenvertreter haben auch die Möglichkeit, die nationale und/oder EU-Gesetzgebung in Sachen Marktzulassungen zu beeinflussen.
Quellenangaben
- The European Economic Area (EEA) unites the EU Member States and the three EEA EFTA States (Iceland, Liechtenstein, and Norway) into the internal market governed by the same basic rules. These rules aim to enable goods, services, capital, and persons to move freely about the EEA in an open and competitive environment, a concept referred to as the four freedoms.
Anlagen
- Marktzulassung
Size: 755,505 bytes, Format: .pptx
Informieren Sie sich über die Verfahren für die Beantragung einer „Genehmigung für das Inverkehrbringen“ (Marktzulassung) eines Arzneimittels im Europäischen Wirtschaftsraum.
A2-5.07-v1.1