Marketing Authorisation Applications
Introduction
The medicines development process is a long journey. The ultimate goal of any development process is an approval for marketing the new medicinal product – a Marketing Authorisation (MA). Within pharmaceutical companies, Regulatory Affairs (RA) departments are (or should be) an integral part of all steps throughout the life-cycle of the medicinal product. Regulatory Affairs are responsible in particular for the applications that must be submitted before every clinical trial, the preparation and submission of the dossier for the Marketing Authorisation Application (MAA), and other activities after the MA is granted, for instance applying for a change to the marketing authorisation (a variation). Regulatory Affairs professionals need to have a thorough knowledge of all applicable medicines regulations and of the entire development process.
-
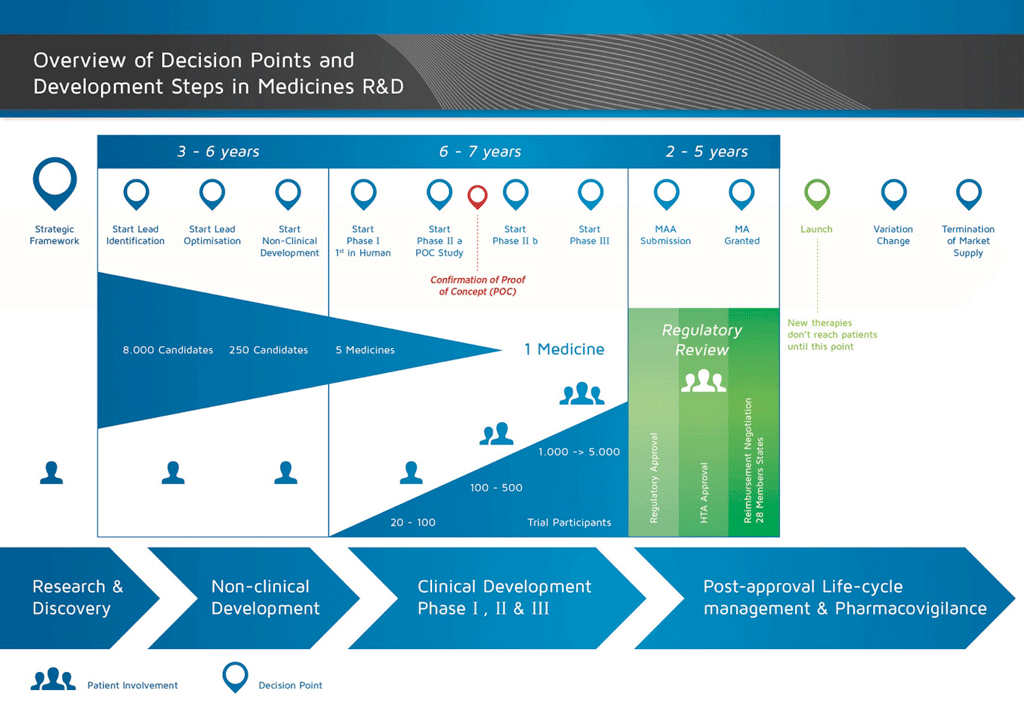
- It takes well over 10 years of careful planning and research for a medicine to go from molecule to a marketable treatment.
Submissions for Marketing Authorisation (MA)
The pharmaceutical company must decide at an early stage of development what type of application to submit for the marketing authorisation e.g.:
- A full application – see Common Technical Document triangle (CTD) below.
- An abridged application (reduced application).
- A bibliographic application – based on existing scientific literature.
Applications require the submission of a dossier of documentation to the relevant authorities. Figure 1 illustrates the development process for a new, innovative medicine. This kind of medicine would require a full dossier to be submitted, in which all elements of documentation for the medicine must be included.
What elements does a dossier include?
Figure 2 shows the elements that make up the Common Technical Document (CTD), the dossier that is submitted to the regulatory authorities as a Marketing Authorisation Application (MAA) in Canada, Europe, Japan, Switzerland, the United States, and others. The format of the CTD has been developed by the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). The CTD is to be used for all types of MAAs in the EU irrespective of the procedure (Centralised Procedure CP, Mutual Recognition Procedure MRP, Decentralised Procedure DCP or National Procedure NP) or the type of application (stand alone, generics, etc.). The CTD format is applicable for all types of products (new chemical entities, radiopharmaceuticals, vaccines, herbals, etc.).
There are five distinct modules in the CTD.
- Module 1: Regional administrative information.
- Module 2: Summaries and overviews.
- Module 3: Quality.
- Module 4: Non-clinical study reports.
- Module 5: Clinical study reports.
Modules 2 to 5 of the CTD are common for all regions, while Module 1 is specific to each region and is not considered part of the CTD. The majority of the documentation relating to the quality, safety, and efficacy of the medicine are in Modules 3 to 5. Regulatory Affairs professionals ensure that the documentation submitted is in-line with all relevant regulations, directives, and guidelines. They also produce the summaries that are included in Module 2.
-
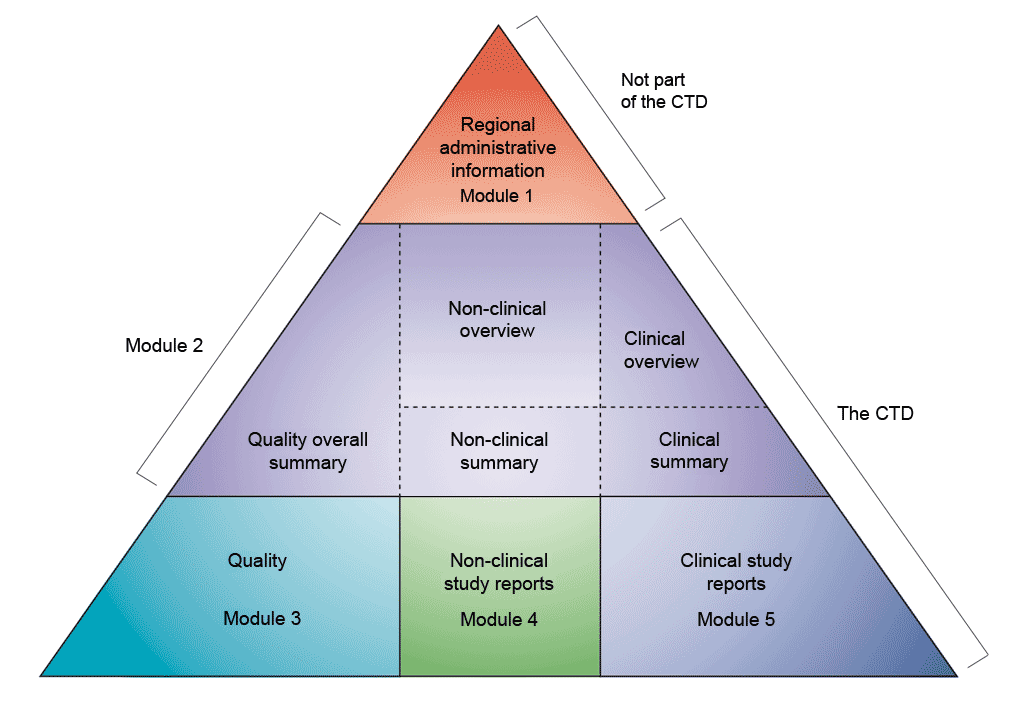
- Diagrammatic representation of the common technical document (CTD) modules. The CTD modules are represented here as a pyramid split into three levels.
Figure 2: The Common Technical Document triangle.
Module 1: Regional administrative information
Module 1 of the CTD contains all the administrative information necessary at a regional level. The EU has its own version of Module 1. It is made up of the following 10 elements:
1.0 Cover letter
1.1 Comprehensive table of contents
1.2 Application form
1.3 Product information
This is information which will be used by both healthcare professionals and patients. It includes the Summary of Product Characteristics (SmPC) – a detailed document for healthcare professionals, labelling and the Package Leaflet (PL). A readability test must be performed to demonstrate that the PL is understandable by lay people. The national competent authorities and the EMA have published templates in all EU languages for the presentation of product information which prescribes the format and content in detail.
1.4 Information about the experts
Module 2 of the CTD contains summaries and overviews written by experts. Each of these experts should provide a curriculum vitae (CV), and must sign a declaration that they have followed the rules of any applicable regulations or directives when creating the summaries.
1.5 Specific requirements for different types of applications
Additional information required for special types of applications, such as Bibliographical Applications, Generic, ‘Hybrid’ or Bio-similar Applications, (Extended) Data / Market Exclusivity, Application under Exceptional Circumstances, or Application for Conditional Marketing Authorisation.
1.6 Environmental risk assessment
All active substances in medicines may pose a potential risk to the environment, and all substances or their metabolites will ultimately end up in the environment. The company must address the possible environmental impact created by the use, storage, and disposal of the medicine.
1.7 Information relating to orphan market exclusivity
Special information is required if the medicine is designated as an orphan medicine intended to treat a rare disease. If another medicine on the market already has market exclusivity for the same indication, the new medicine may only be approved under special conditions.
1.8 Information relating to pharmacovigilance
A description of the pharmacovigilance and risk management systems must be included. The applicant must demonstrate that proper surveillance of adverse reactions and potential risks is in place. This should include proof that the applicant has a qualified person responsible for pharmacovigilance and the necessary means for the notification of any adverse reaction occurring either in the EU or in a third country (Article 8 (n) of Directive 2001/83/EC).
1.9 Information relating to clinical trials
The applicant must include a statement that any clinical trials of the medicine performed outside of the EU met EU requirements.
1.10 Information relating to paediatrics
In the EU, all new medicines must be considered for the paediatric population. In general, they should be tested in children. However, a waiver may be granted for this requirement if the disease is only seen in elderly people or adults, or if the new medicine is likely to be ineffective or unsafe in part or all of the paediatric population. If a waiver is not granted, the company must produce a Paediatric Investigation Plan (PIP) – unless a deferral has been granted, in which case the PIP may be produced later. This section should include a copy of the waiver or of the decision on the PIP (including deferrals, if applicable).
How is the dossier put together?
In most cases, paper submissions are no longer possible, meaning that all the documentation within the five CTD modules should be in a standardised electronic format: the eCTD. The eCTD is not just a collection of PDF documents, nor a single huge PDF file. Rather, the eCTD is a standard that describes in detail the folder and file structure that must be followed so that the company and the regulatory authorities can easily navigate the dossier, just as you would in a normal computer directory.
What is the submission process?
The applicant must carefully consider all logistical and regulatory issues prior to submission. This includes choosing which marketing authorisation procedure to pursue: the Centralised Procedure (CP), the Mutual Recognition Procedure (MRP), the Decentralised Procedure (DCP), or the National Procedure (NP).
-
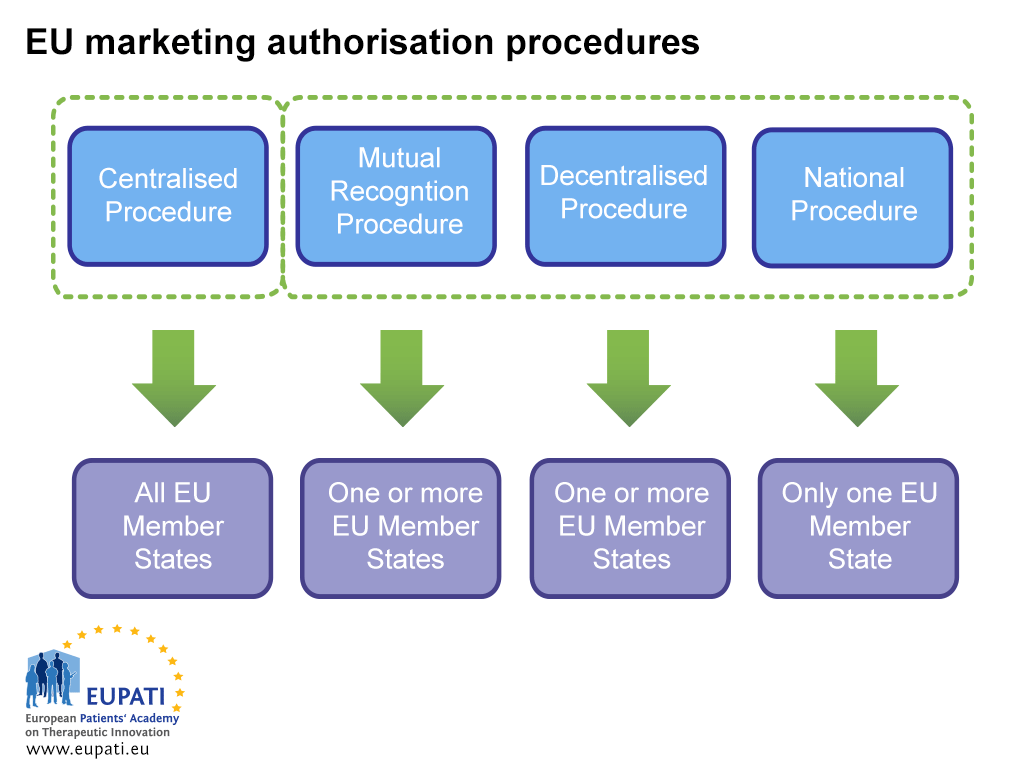
- Different actors are involved in the marketing authorisation of a medicine depending on which procedure the sponsor opts (or is obliged) to follow.
Answers to many questions related to applications via the CP may be found on the EMA website.
Pre-submission meetings
Pre-submission meetings between the company and regulatory staff generally take place six to seven months before the submission date. These meetings are arranged so that the company can seek further information and guidance before finalising the application dossier.
For MAAs following the CP, the project team from the company will meet with the team from the EMA that will be involved in assessing the application. In MRP, DCP, or NP, pre-submission meetings with the relevant national competent authorities are possible and equally useful.
Submission of the Marketing Authorisation Application (MAA)
In the CP, submissions are only possible to the EMA in eCTD format, unless an exception is granted. The eCTD is submitted through an online portal.
In the case of applications following the MRP, DCP, or NPs, the situation is more complicated. These applications may involve up to 31 different agencies. The HMA network (Heads of Medicines Agencies network, a collaboration between all Member States) now offers a similar solution to the EMA: the Common European Submission Platform (CESP). Using the CESP means that the company only needs to upload a dossier into the system once; all the Member States involved can then extract the submission form the CESP repository. The platform also allows for communication between agencies and the applicant.
The validation phase
When the EMA or the national competent authority receives the MAA submission, the dossier is first validated to ensure that all the necessary documentation has been included. Where there are questions the applicant is given the opportunity to supply the necessary responses and supporting documentation. Once the MAA is validated, the assessment begins.
A2-5.11-V1.1