Icke-klinisk utveckling:grundläggande principer
Inledning
Den icke-kliniska (eller prekliniska) utvecklingsfasen syftar huvudsakligen till att identifiera vilken kandidatbehandling som har störst sannolikhet att bli framgångsrik, bedöma dess säkerhet och bygga en stabil vetenskaplig grund innan man övergår till den kliniska utvecklingsfasen.
Under den icke-kliniska utvecklingsfasen ska kandidatföreningen även uppfylla icke-medicinska mål, bland annat ska man definiera de immateriella rättigheterna och se till att en tillräcklig mängd av läkemedelsprodukten finns tillgänglig för kliniska prövningar.Den icke-kliniska utvecklingen av ett läkemedel är komplex och styrs av regelverk.
Grundläggande information, viktiga definitioner och begrepp
”Icke-klinisk” eller ”preklinisk”?
Termerna ”icke-klinisk” och ”preklinisk” används ofta omväxlande.
Även om icke-kliniska studier har avgörande relevans under de prekliniska utvecklingsstegen kan de utföras när som helst under produktens livscykel, men i många fall är det bättre att utföra dem så tidigt som möjligt för att undvika överraskningar senare under utvecklingen.
Data från icke-kliniska studier används för att identifiera farmakodynamiken (hur läkemedlet påverkar kroppen), farmakokinetiken (hur kroppen påverkar läkemedlet) och toxikologin hos kandidatföreningen innan den ges till människor. Därutöver används de för att förfina, befästa och lägga till information för att uppdatera produktens säkerhetsprofil under den prekliniska fasen, vid tidpunkten för registrering och under läkemedelsproduktens livscykel.
Studier in silico, in vitro och in vivo
Under icke-klinisk utveckling utförs studier
- in silico:på en dator eller via datorsimulering, t.ex. för att förutsäga den toxikologiska profilen för en produkt med hjälp av dess kemiska struktur genom databaserade metoder
- in vitro (latin för ”i glas”):utförande av en procedur i en kontrollerad miljö utanför en levande organism, till exempel med hjälp av hepatocytodlingar (odlingar av celler från levern) för metabolismstudier
- in vivo (latin för ”i det levande”):försök med en hel levande organism i motsats till vävnader eller celler, dvs. djur, människor eller växter.
Vilka är de viktigaste aspekterna av kemi, tillverkning, kontroll (CMC) under icke-klinisk utveckling?
För alla icke-kliniska utvecklingsstudier krävs tillverkning av en lämplig aktiv substans:
- För icke-kliniska studier behövs vanligtvis små mängder (milligram till gram). Sedan måste en uppskalningsprocess utvecklas för att producera större mängder för kliniska prövningar och i ett senare skede, efter godkännande, för marknaden.
- För studier enligt god laboratoriesed (GLP) krävs godkända partier eller partier tillverkade enligt god tillverkningssed (GMP) av den aktiva substansen.
Här följer några viktiga åtgärder beträffande kemi, tillverkning och kontroll (CMC) under den icke-kliniska utvecklingsfasen:
- bestämning av dos och tillförsel
- detaljerad fysikalisk-kemisk karakterisering
- stabilitetstestning och föroreningsanalys
- utveckling och validering av metoder för att bestämma mängden av den aktiva substansen i kroppsvätskor som blod, plasma och urin i aktivitets- och biverkningsstudier
- utveckling av en prototyp för klinisk användning.
Den icke-kliniska utvecklingsprocessen
Icke-kliniska utvecklingsaktiviteter sker parallellt med forskningsaktiviteterna.De ska stödja det planerade kliniska utvecklingsprogrammet genom att arbeta mot de mål och besvara de frågor som beskrivs nedan.
Mål
När en kandidatförening har identifierats ska man under den icke-kliniska utvecklingen börja med att besvara följande frågor, vars svar kommer från specifika utvärderingar/studier:
- Fungerar den?→ bedömning av effekt
- Hur ska den tillföras och hur reagerar kroppen?→ profilering
- Är den säker?→ toxikologi/säkerhet
- Är tillverkningen genomförbar och kontrollerbar?
Icke-kliniska utvecklingsaktiviteter kan fortsätta genom produktens hela livscykel, men ju tidigare dessa frågor besvaras, desto lättare är det att identifiera profilen på den patient som har den största nyttan.
Projekthantering
Det icke-kliniska utvecklingsprogrammet är komplext och kräver gedigna färdigheter i projekthantering och kommunikation när det gäller att leda multidisciplinära team.Projektteamet måste förstå den avsedda kliniska planen för att kunna definiera den icke-kliniska planen och tillhörande aktiviteter.
Profilen ger ett ramverk för att verkställa den icke-kliniska utvecklingsstrategin, definiera mål, risker, ansvar och mätningar samt fatta beslut om att fortsätta/avsluta.Profilimplementering bidrar till att rikta in projektets fokus mot viktiga produktkriterier, fatta beslut om att fortsätta/avsluta i rätt tid och minska den totala risken för projektet (t.ex. fortsatt utveckling av en oanvändbar produkt).
Icke-kliniska reglerande riktlinjer
Det är många aktörer inblandade i utvecklingen av läkemedel och varje organisation eller institution följer sina egna regler.Exempelvis har företag olika standardrutiner (SOP).Utöver bestämmelser om god klinisk sed finns information om riktlinjer på Europeiska läkemedelsmyndighetens (EMA:s) webbplats.
- De är antingen allmänna eller mer specifika och behandlar vetenskapliga och tekniska aspekter (t.ex. specifika för nödvändiga toxikologistudier).
- De måste följas strikt vid ansökan om ett nytt godkännande för försäljning. Eventuella avvikelser måste motiveras.
Data presenteras i CTD-formatet (gemensamt tekniskt dokument) som definierats av den internationella konferensen om harmonisering av tekniska krav för registrering av humanläkemedel (ICH).Överenskommelsen om att samla all information om kvalitet, säkerhet och effekt i detta gemensamma format (CTD) har revolutionerat processen för myndighetsgranskning och lett till harmoniserad elektronisk inlämning som i sin tur möjliggör införande av god granskningssed.För branschen har det inneburit att informationsformatet för inlämning till de olika tillsynsmyndigheterna inte behöver ändras (ICH sammanför tillsynsmyndigheterna och läkemedelsindustrin i Europa, Japan och USA för att diskutera vetenskapliga och tekniska aspekter på läkemedelsregistrering).
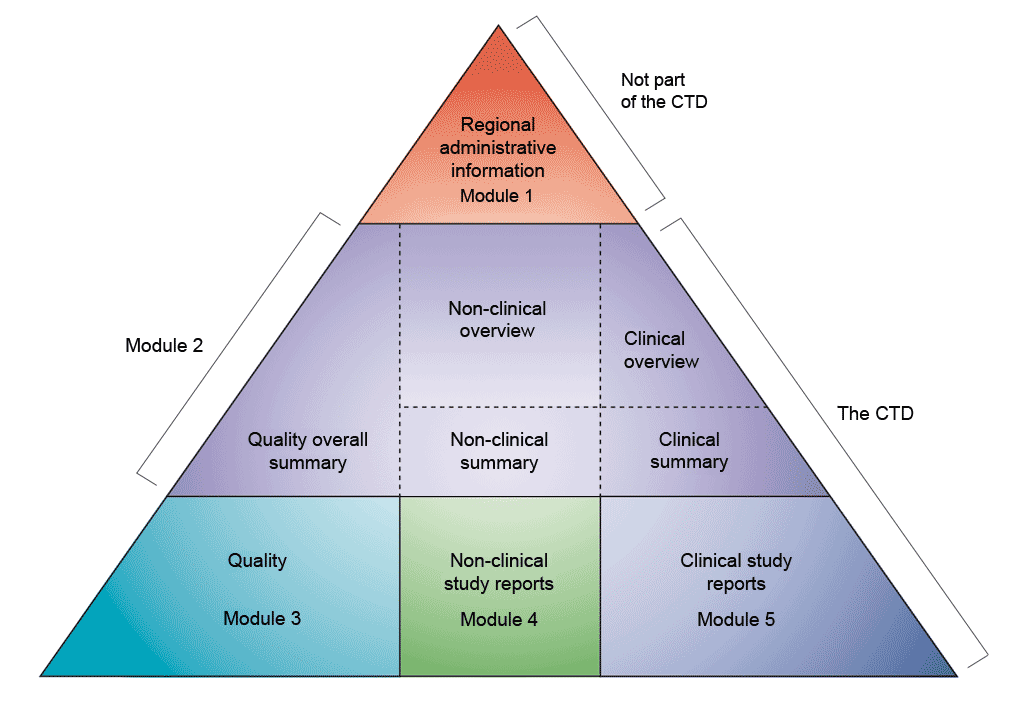
CTD-dokumentet är indelat i fem moduler (se bilden ovan).I juli 2003 blev CTD det obligatoriska formatet för nya ansökningar om godkännande för försäljning i EU och Japan och det starkt rekommenderade formatet för nya läkemedelsansökningar (NDA) till den amerikanska livsmedels- och läkemedelsmyndigheten (Food and Drug Administration; FDA).
Sammanfattning
Den icke-kliniska utvecklingsfasen är kritisk och under denna fas måste potentiella problem förutses innan en förening överförs till den kliniska utvecklingsfasen.
För att kliniska studier av en kandidatförening ska tillåtas krävs följande:
- Icke-klinisk säkerhetsutvärdering ska ha utförts under förhållanden med god laboratoriesed (GLP).
- Tillverkning ska ha utförts under lämplig kvalitetskontroll.
- Data och process ska vara dokumenterade i enlighet med CTD-formatet och man ska ha byggt en grund för den kliniska utvecklingsfasen.
Det finns en ökad tendens att utforma läkemedelslika egenskaper in silico samt att använda bioinformatiska metoder för modellering och förutsägelser.
Riktlinjer för icke-klinisk utveckling är föremål för kontinuerlig harmonisering mellan de viktigaste tillsynsmyndigheterna (Europa, USA och Japan) som framhåller säkerhet och kvalitet:ICH ger regelbundet ut detaljerade riktlinjer för läkemedelsindustrin liknande de som publiceras av instanser i Europa (EMA) och USA (FDA).
Ytterligare resurser
- European Medicines Agency (2007a). Guideline on strategies to identify and mitigate risks for first-in human trials with investigational medicinal products. London: European Medicines Agency. Retrieved 25 June, 2015, from http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002988.pdf
- European Medicines Agency (2007b). Guideline on requirements for first-in-man clinical trials for potential high-risk medicinal Retrieved 25 June, 2015, from http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002989.pdf
- European Medicines Agency (2009). ICH guideline M3(R2) on non-clinical safety studies for the conduct of human clinical trials and marketing authorisation for pharmaceuticals. London: European Medicines Agency. Retrieved 25 June, 2015, from http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002720.pdf
- European Medicines Agency (2011). Committee for medicinal products for human use (CHMP) ICH guidelines S6 (R1) – preclinical safety evaluation of biotechnology-derived pharmaceuticals. London: European Medicines Agency. Retrieved 25 June, 2015, from http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002828.pdf
[glossary_exclude]Referenser
- Image reproduced from ICH (2015). M4: The Common Technical Document. Retrieved 11 July, 2021, from https://www.ich.org/page/ctd
- European Medicines Agency (2009). ICH guideline M3(R2) on non-clinical safety studies for the conduct of human clinical trials and marketing authorisation for pharmaceuticals. London: European Medicines Agency. Retrieved 25 June, 2015, from http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002720.pdf[/glossary_exclude]
A2-2.01.1-1.2