Grundlagen der Arzneimittelregulierung
Warum werden Arzneimittel reguliert?
Jeder Mensch möchte im Falle einer Krankheit die Möglichkeit einer medizinischen Behandlung haben. Dafür werden Arzneimittel gebraucht, die gegen die Krankheit wirksam sind.
Unglücklicherweise haben alle Arzneimittel auch unerwünschte Wirkungen. Dennoch müssen alle auf dem Markt erhältlichen Arzneimittel in der normalen Anwendung sicher sein.
Arzneimittel müssen zuverlässig sein. Das bedeutet, dass sie nach hohen Qualitätsstandards hergestellt werden müssen.
Mit allen diesen Gesichtspunkten befasst sich Regulierung. Arzneimittel werden reguliert, um zu gewährleisten, dass nur ausreichend sichere, wirksame und qualitativ hochwertige Arzneimittel auf den Markt kommen.
Wer reguliert Arzneimittel weltweit?
Es gibt keine weltweite Regulierung von Arzneimitteln. Seit über 20 Jahren gibt es jedoch Bemühungen zur weltweiten Harmonisierung der Arzneimittelregulierung. Im International Council on Harmonisation (ICH, Internationaler Rat für die Harmonisierung) arbeiten Zulassungsbehörden und die pharmazeutische Industrie aus der EU, den USA, Japan, Kanada, der Schweiz und andere nationale Organisationen zusammen, und die Weltgesundheitsorganisation (WHO) sowie einige nationale legislative oder administrative Behörden fungieren als Beobachter.
Wer reguliert Arzneimittel in der Europäischen Union (EU)?
In der EU wird die Arzneimittelregulierung zwischen allen Mitgliedstaaten unter einem gemeinsamen gesetzlichen Regelwerk harmonisiert. Akteure sind die Europäische Kommission (EC), die Europäische Arzneimittel-Agentur (EMA) und die nationalen Aufsichtsbehörden, auch Zulassungsbehörden genannt. Dieses Regelwerk erstreckt sich auch auf den Europäischen Wirtschaftsraum (EWR), zu dem auch Norwegen, Island und Liechtenstein gehören. In der restlichen Welt werden Arzneimittel auf nationaler Ebene durch die Aufsichtsbehörden der jeweiligen Länder reguliert, und diese Arbeit wird innerhalb der ICH-Regionen harmonisiert.
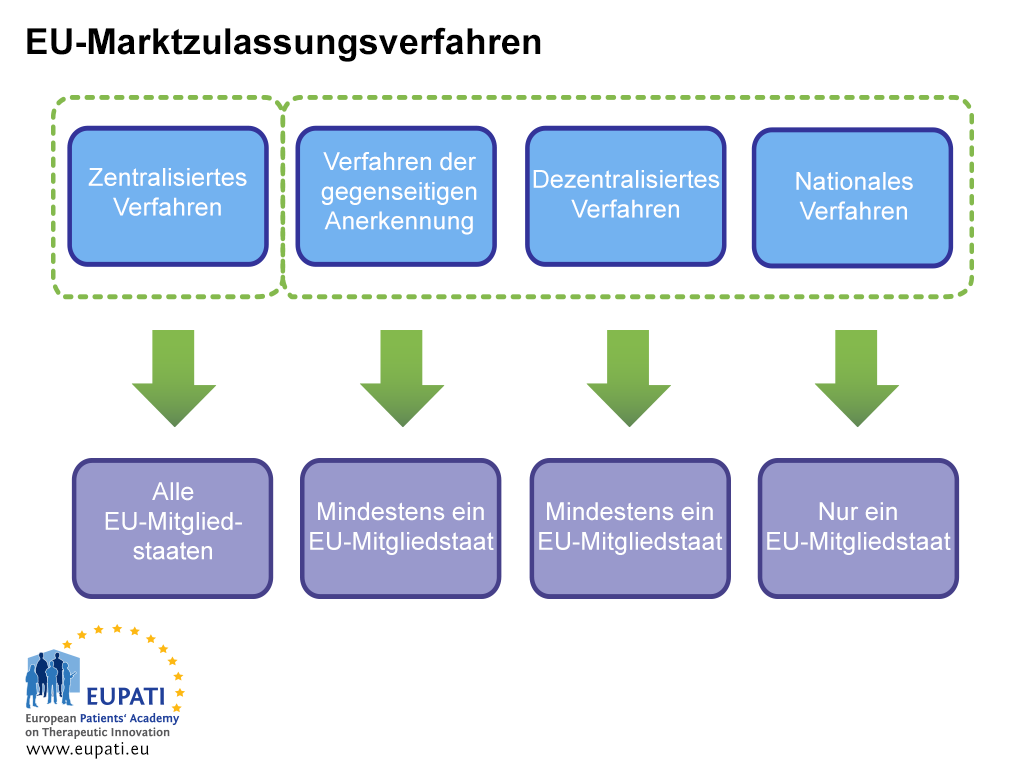
Aufgrund dieser Harmonisierung können Arzneimittel in der EU gleichzeitig in allen EU- und EWR-Ländern zugelassen werden. Diese geschieht mithilfe des Zentralisierten Verfahrens, das von der EMA beaufsichtigt wird. Ein Arzneimittel kann in den EU-Mitgliedstaaten auch über das Dezentralisierte Verfahren, das Verfahren der gegenseitigen Anerkennung und im nationalen Verfahren zugelassen werden. Bei diesen Verfahren sind nationale Aufsichtsbehörden zuständig, und sie gelten nicht automatisch für alle europäischen Mitgliedstaaten.
-
- An der Marktzulassung eines Arzneimittels sind verschiedene Parteien beteiligt, je nachdem, welches Verfahren der Sponsor für die Beantragung ausgewählt hat (oder befolgen muss).
Die Rolle der Europäischen Arzneimittel-Agentur in der Regulierung und Zulassung von Arzneimitteln
Die Europäische Arzneimittel-Agentur (EMA) spielt eine wichtige Rolle sowohl in der Regulierung als auch in der Zulassung von Arzneimitteln.
Die Europäische Arzneimittel-Agentur trifft ihre Entscheidungen über die Zulassung von Arzneimitteln auf der Basis von klinischen Studien, die von pharmazeutischen Firmen durchgeführt werden. Sie unterhält außerdem eine Datenbank der klinischen Studien, die in der Europäischen Union durchgeführt werden: https://www.clinicaltrialsregister.eu/.
Die EMA überwacht das Zentralisierte Verfahren für die Marktzulassung. Dies ist das Verfahren, in dem die meisten Zulassungsanträge für Arzneimittel gestellt werden. Das betreffende Unternehmen muss der EMA dazu einen einzigen Antrag vorlegen. Wenn diese zu einer positiven Entscheidung kommt, erhält das Arzneimittel von der EC die Genehmigung für das Inverkehrbringen in allen EU- und EWR-Ländern.
Innerhalb der EMA ist der Ausschuss für Humanarzneimittel (CHMP) für die Begutachtung der Dossiers (Anträge) zuständig. Die Sachverständigen, die die Anträge begutachten, werden von den einzelnen Mitgliedstaaten und zusätzlich von Island und Norwegen ernannt. Der Ausschuss kann, wenn nötig, unter den von den Mitgliedstaaten oder der EMA benannten Sachverständigen bis zu fünf zusätzliche Mitglieder in den Ausschuss berufen („kooptieren“), um zusätzliche Kompetenz in einem bestimmen wissenschaftlichen Feld im Team zu haben.
A2-5.01.1-V1.1