Galenisk formulering: Sådan er lægemidler sammensat (formuleret)
Galenisk farmaci er processen, hvor et aktivt stof omdannes til et brugsklart lægemiddel, som kan doseres efter behov. Galenisk formulering handler om principperne for forberedelse og sammensætning af lægemidler forat optimere deres absorption og er led i farmaci, disciplinen (eller videnskaben) for design af doseringsform.
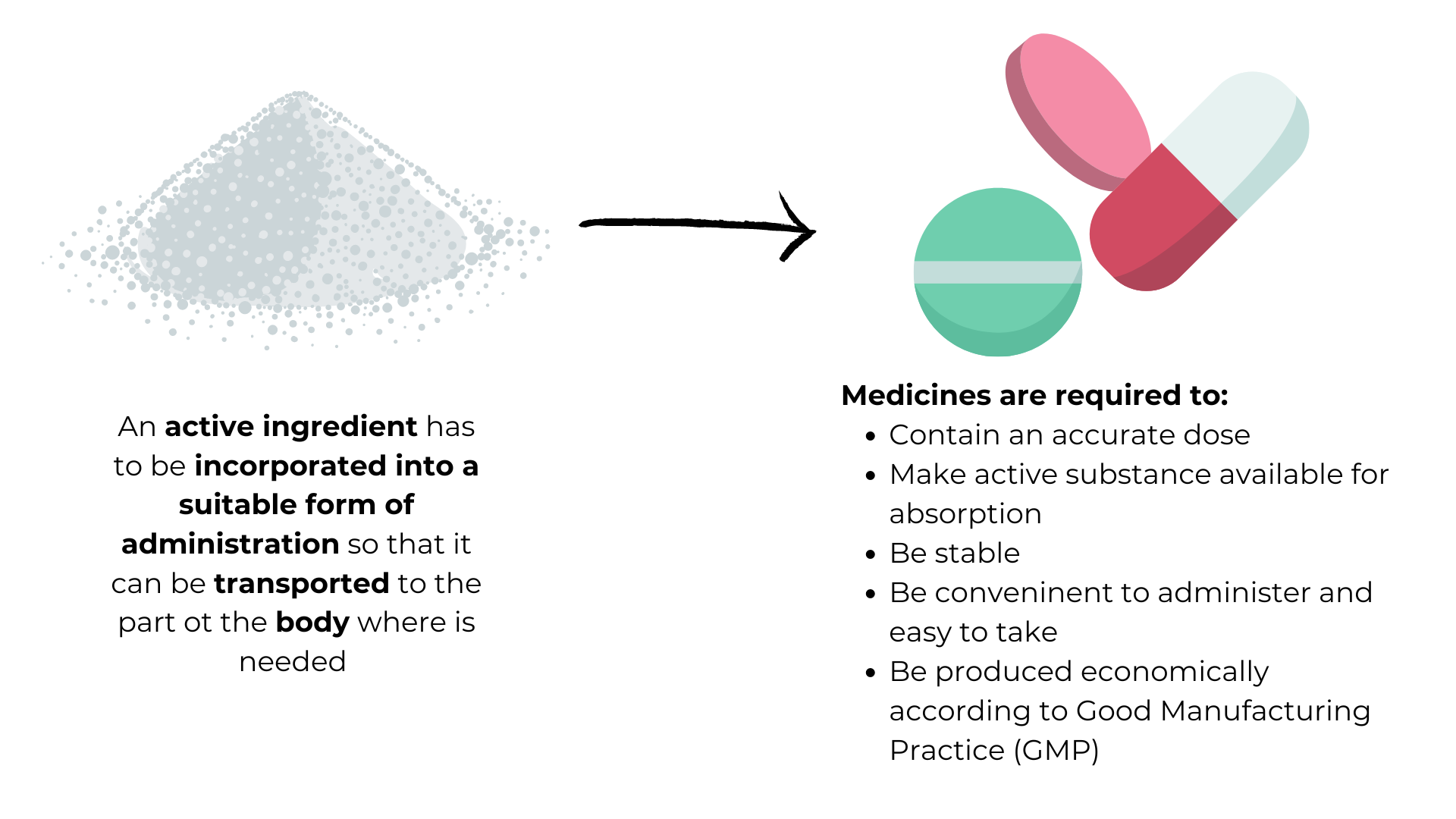 Der kræves et aktivt stof til udvikling af formulering, undersøgelser af sikkerhedsvurderinger og kliniske undersøgelser. Der produceres en tilstrækkelig mængde aktivt stof af høj kvalitet i en kemisk udviklingsproces, som skal bruges til sikkerhedstest og af udviklere til at udforme et lægemiddel.
Der kræves et aktivt stof til udvikling af formulering, undersøgelser af sikkerhedsvurderinger og kliniske undersøgelser. Der produceres en tilstrækkelig mængde aktivt stof af høj kvalitet i en kemisk udviklingsproces, som skal bruges til sikkerhedstest og af udviklere til at udforme et lægemiddel.
Lægemidlet består af formuleringen af det aktive stof (tablet, creme, suspension,opløsning), hjælpestoffer (inaktive indholdsstoffer som laktose) og emballage/leveringsanordning (blisterpakke, flaske, inhalator, hætteglas, fyldt injektionssprøjte).
Galenisk formulering til prækliniske undersøgelser
Prækliniske sikkerhedsundersøgelsers har til formål at:
- undersøge responsen op til det maksimalt tilladelige antal doser
- opdage potentielle skadevirkninger
- generere tilstrækkelige data til at foretage en risikovurdering
- medvirke til dosisvalg for indledende kliniske undersøgelser
- foreslå “markører” for at overvåge sikkerheden i mennesker
- danne grundlag for målrettede specialiserede undersøgelser
Disse sikkerhedsundersøgelser kan dog ikke nødvendigvis garantere sikkerheden for mennesker eller forudsige menneskers respons. Sikkerhedstest undersøger sikkerhedsmarginer i data fra dyr og mennesker. I forbindelse med disse undersøgelser skal følgende overvejes:
- indgivet dosis,
- omfang og varighed af systemisk eksponering (tilstedeværelse i kroppen),
- daglig systemisk eksponering,
- eksponering og identitet af metabolitter (nedbrydningsprodukter) og
- eksponering i målorganer.
Sikkerhedstest udføres på det aktive stof samt på alle relaterede stoffer, opløsningsmidler, nedbrydningsprodukter, hjælpestoffer og andre aktive materialer og ekstraktstoffer, som er en del af den endelige formulering.
Galenisk formulering til kliniske undersøgelser
Faktorer, der skal tages højde for i udviklingen af en egnet administrationsform, omfatter patientens accept af administrationsformen og specifikke krav for det aktive stof. Inaktive indholdsstoffer eller hjælpestoffer kan udgøre det meste af lægemidlets volumen og fungere som bærere af det aktive stof. Majsstivelse eller laktose bruges for eksempel i tabletter, mens emulsioner bestående af vand og olie bruges i salver.
Administrationsformen påvirker også absorption (optagelse), tilgængelighed af det aktive stof og derfor et lægemiddels terapeutiske effekt. Den afgør, hvordan det aktive stof kommer ind i kroppen, hvor og i hvilken dosis det frigives, samt den tid det tager at blive absorberet. Desuden skal administrationsformen sikre, at patienten kan dosere lægemidlet sikkert og nemt håndtere det.
Forskere i formulering sørger for, at stoffet kan absorberes af kroppen, og at behandlingsdosis når frem til det ønskede organ. Ikke alle aktive stoffer er egnet til indgivelse i tabletform, og særlige krav til administrationsformen (injektion i øjet, produkter til inhalation eller tabletter, som opløses i patientens mund) skaber jævnligt nye udfordringer for forskere i formler.
God fremstillingspraksis (GFP)
Alle lægemidler, herunder generiske lægemidler og biosimilære midler, skal fremstilles i henhold til retningslinjer for god fremstillingspraksis (GFP), inden de godkendes til mennesker.
Retningslinjerne for GFP er en vejledning i fremstilling, test og kvalitetssikring med henblik på at sikre, at et lægemiddel er sikkert for mennesker at indtage. Mange lande har specifikke love og bestemmelser, som pålægger producenter af farmaceutisk og medicinsk udstyr at følge GFP-procedurerne. De fleste bestemmelser er baseret på internationalt aftalte retningslinjer.
Retningslinjerne for GFP følger nogle få grundlæggende principper:
- Hygiejne: Farmaceutiske produktionsanlæg skal være rene og hygiejniske.
- Kontrollerede miljømæssige betingelser for at forhindre krydskontaminering af et lægemiddel fra et andet stof eller ikke-relaterede partikler, hvilket kan gøre lægemidlet usikkert at indtage for mennesker.
- Produktionsprocesser klart definerede og kontrollerede:
- Alle kritiske processer kontrolleres for at sikre konsistens og overensstemmelse med specifikationer.
- Alle ændringer af processen evalueres.
- Ændringer, der har indflydelse på kvaliteten af lægemidlet, valideres efter behov.
- Instruktioner og procedurer er skrevet i et klart og utvetydigt sprog. (Gode dokumentationspraksis – GDP).
- Operatører uddannes til at udføre og dokumentere procedurer.
- Journaler genereres manuelt eller af instrumenter under fremstillingen for at påvise, at alle de trin, der kræves for de definerede procedurer og instruktioner, faktisk er udført, og at mængden og kvaliteten af lægemidlet er som forventet.
- Afvigelser undersøges og dokumenteres.
- Dokumenter for fremstilling (inklusive distribution), der beskriver den komplette historie for et parti, så det kan spores, opbevares i et forståeligt og tilgængeligt format.
- Distribution af lægemidlerne minimerer enhver risiko for at forringe kvaliteten.
- Der er etableret et system til tilbagekaldelse af et parti af lægemidlet fra salg eller levering.
- Klager over markedsførte lægemidler undersøges, årsagerne til mangelfuld kvalitet undersøges, og der træffes hensigtsmæssige foranstaltninger i forhold til de mangelfulde lægemidler og for at forhindre, at det sker igen.
Flere ressourcer
A2-2.06-V1.4