Galenic formulation: How medicines are formulated
Galenics is the process that turns an active ingredient into a ready-to-use medicine that can be dosed as required. Galenic formulation deals with the principles of preparing and compounding medicines inorder to optimise their absorption and forms part of pharmaceutics, the discipline (or science) of dosage form design.
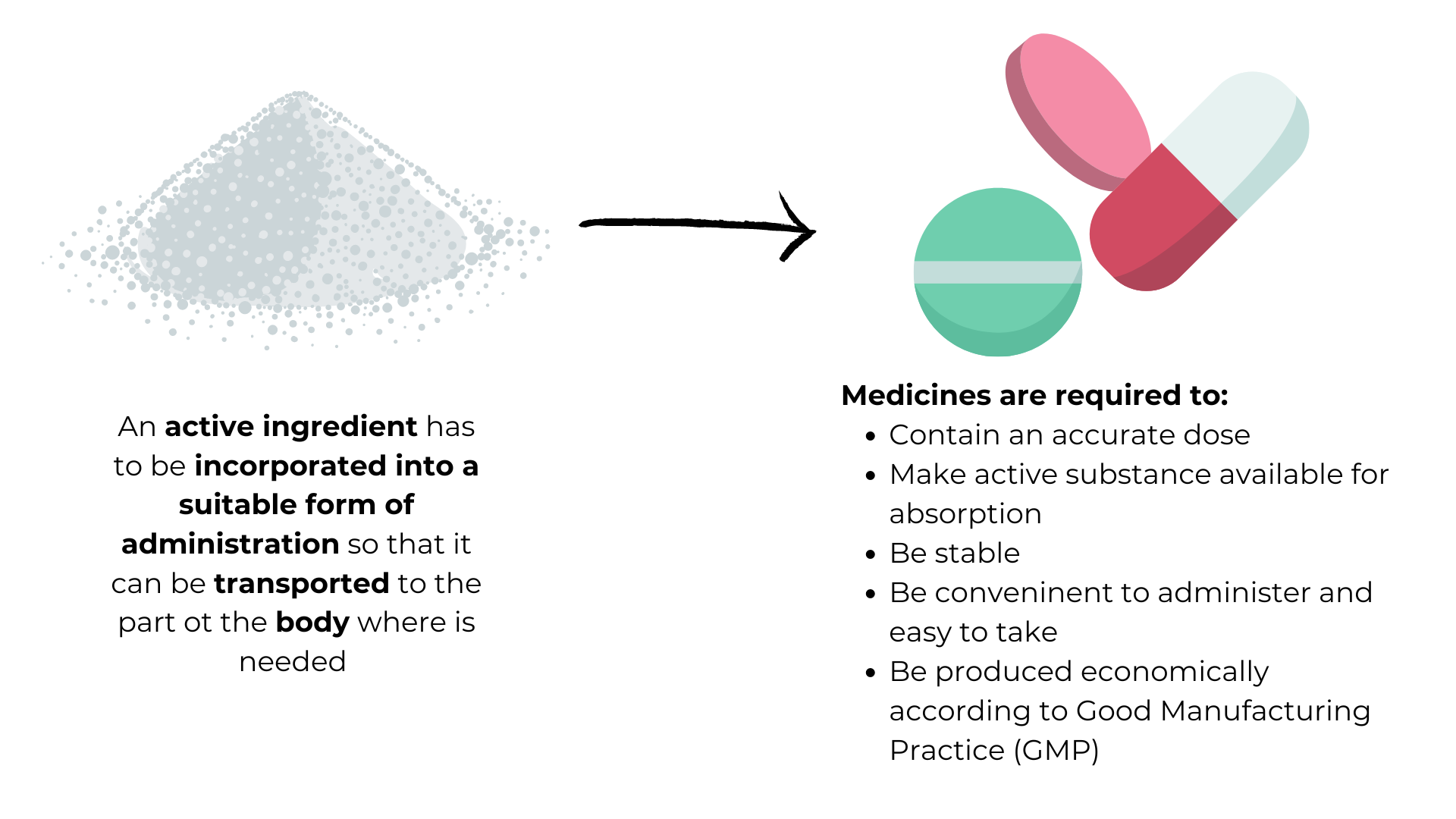 Diagrammatic representation of galenic formulation – the incorporation of an active ingredient into finished medicines ready for administration – and the requirements that those medicines must fulfil.
Diagrammatic representation of galenic formulation – the incorporation of an active ingredient into finished medicines ready for administration – and the requirements that those medicines must fulfil.
An active substance is required for formulation development, safety assessment studies, and clinical studies. A sufficient quantity of quality active substance is produced in a chemical development process to be used in safety testing and for scientists to formulate a medicinal product. The medicinal product will comprise the active substance formulation (tablet, cream, suspension,solution), excipients (inactive ingredients such as lactose), and packaging/delivery device (blisterpack, bottle, inhaler, vial, prefilled syringe).
Galenic formulation for non-clinical studies
Non-clinical safety studies aim to:
- Explore the response at up to maximum tolerable doses
- Detect potential hazards
- Generate enough data to make a risk assessment
- Assist in dose selection for initial clinical studies
- Suggest ‘markers’ to monitor safety in humans
- Provide a foundation for targeted specialised investigations
However, these safety studies cannot necessarily guarantee safety in humans or predict human responses. Safety testing assesses the margins of safety in animal and human data. Considerations for these types of tests are:
- dose administered,
- extent and duration of systemic exposure,
- daily systemic exposure,
- exposure and identity of metabolites, and
- exposure in target organs.
Safety testing is carried out on the active substance, as well as on all related substances, solvents, degradation products, excipients, and other active materials and extractives that are part of the final formulation.
Galenic formulation for clinical studies
Factors to be considered in the development of a suitable form of administration include patient acceptability and specific requirements of the active ingredient. Inactive ingredients or excipients can make up most of a medicine’s volume and serve as carriers for the active compound. Maize starch or lactose, for example, are used in tablets, while water-oil emulsions are used in ointments. The administration form also influences absorption, availability of the active substance, and therefore a medicine’s therapeutic effect. It determines how the active ingredient enters the body, where and in what dosage is released, and the time it takes to be absorbed. In addition, the mode of administration must ensure that the patient will be able to dose the medicine safely and handle it easily. Formulation scientists ensure that the substance can be absorbed by the body and that the therapeutic dose reaches the targeted organ. Not every active substance is suitable for ingestion as a tablet, and special demands on the form of administration (injection into the eye, products for inhalation, or tablets that dissolve in the patient’s mouth) regularly create new challenges for formulation scientists.
Good Manufacturing Practice (GMP)
All medicines, including generic medicines and biosimilars, should be produced according to Good Manufacturing Practice (GMP) guidelines before being administered to humans. The GMP guidelines provide direction for manufacturing, testing, and quality assurance, in order to ensure that a medicine is safe for human consumption. Many countries have specific laws and regulations imposing that pharmaceutical and medical device manufacturers follow GMP procedures. Most regulations are based on internationally agreed guidelines. The GMP guidelines follow a few basic principles:
- Hygiene: Pharmaceutical manufacturing facilities must maintain a clean and hygienic area.
- Controlled environmental conditions in order to prevent cross contamination of a medicinal product from other compound or unrelated particulate matter, which may make the medicine unsafe for human consumption.
- Manufacturing processes clearly defined and controlled:
- All critical processes are validated to ensure consistency and compliance with specifications.
- Any changes to the process are evaluated.
- Changes that have an impact on the quality of the medicine are validated as necessary.
- Instructions and procedures are written in clear and unambiguous language. (Good Documentation Practices – GDP).
- Operators are trained to carry out and document procedures.
- Records are generated manually or by instruments during manufacture to demonstrate that all the steps required by the defined procedures and instructions were actually taken, and that the quantity and quality of the medicine is as expected.
- Deviations are investigated and documented.
- Records of manufacture (including distribution) that enable the complete history of a batch to be traced are retained in a comprehensible and accessible form.
- The distribution of the medicines minimizes any risk to their quality.
- A system is available for recalling any batch of medicine from sale or supply.
- Complaints about marketed medicines are examined, the causes of quality defects are investigated, and appropriate measures are taken with respect to the defective medicines and to prevent recurrence.
Further resources
A2-2.06-V1.4