Fondamenti della regolamentazione dei farmaci
Perché i farmaci vengono regolamentati?
Tutti desiderano essere in grado di ricevere un trattamento medico se sono ammalati. Sono perciò necessari medicinali che siano efficaci contro la malattia.
Sfortunatamente tutti i farmaci hanno effetti collaterali indesiderati. Indipendentemente da questo, i medicinali sul mercato devono essere sicuri per un uso normale.
I farmaci devono essere affidabili. Ciò significa che devono essere fabbricati secondo standard di elevata qualità.
Tutti questi punti vengono affrontati mediante una regolamentazione. I medicinali vengono regolamentati per assicurare che siano immessi in commercio solo farmaci sufficientemente sicuri, efficaci e di elevata qualità.
Chi regola i farmaci a livello globale?
Non esiste una regolamentazione dei farmaci a livello globale. Tuttavia, per più di 20 anni, sono stati fatto sforzi per armonizzare le normative relative ai farmaci a livello globale. Il Consiglio internazionale sull’armonizzazione (International Council on Harmonisation, ICH) comporta la collaborazione tra gli enti di regolamentazione e l’industria farmaceutica dell’UE, degli Stati Uniti, di Giappone, Canada, Svizzera e altre organizzazioni regionali, con l’Organizzazione mondiale della sanità (World Health Organisation, WHO) e un numero di autorità normative o amministrative nazionali che agiscono come osservatori.
Chi regola i medicinali nell’Unione Europea (UE)?
Nell’UE, la regolamentazione dei medicinali viene armonizzata tra tutti gli Stati membri sotto un insieme comune di norme legislative, e comprende la Commissione europea (CE), l’Agenzia europea per i medicinali (EMA) e le autorità nazionali competenti (National Competent Authorities, NCA) (enti di regolamentazione). Tali norme coprono anche lo Spazio economico europeo (SEE), che include Norvegia, Islanda e Liechtenstein. Nel resto del mondo i farmaci sono regolamentati a livello nazionale dall’autorità nazionale competente (NCA) di ogni paese e armonizzati nelle regioni dell’ICH.
A causa di quest’armonizzazione, i farmaci nell’UE possono essere autorizzati contemporaneamente in tutti i paesi dell’UE e dello SEE mediante la procedura centralizzata, la quale viene supervisionata dall’EMA. Un farmaco può essere inoltre autorizzato negli Stati membri dell’UE tramite la procedura decentralizzata (Decentralised Procedure, DCP), la procedura di mutuo riconoscimento (Mutual Recognition Procedure, MRP) e la procedura nazionale. Queste coinvolgono le autorità nazionali competenti (NCA) e non si applicano automaticamente a tutti gli Stati membri dell’Europa.
-
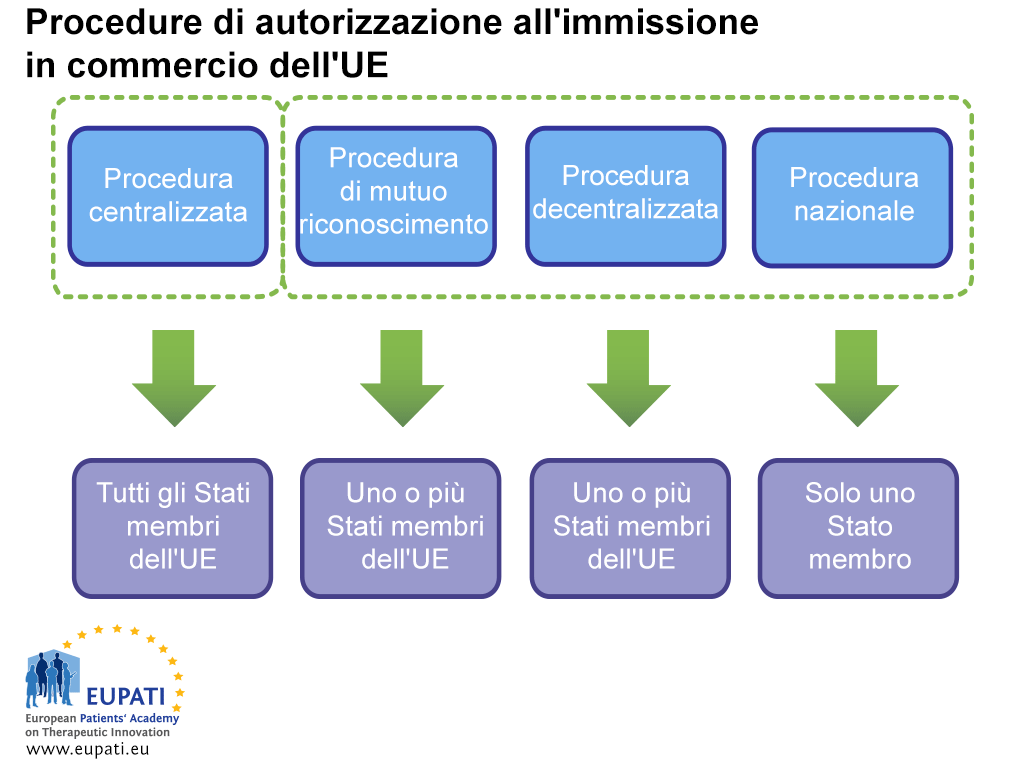
- In funzione della procedura che lo sponsor sceglie o è obbligato a seguire, vi sono diverse parti coinvolte nell’autorizzazione all’immissione in commercio di un farmaco.
Il ruolo dell'Agenzia europea per i medicinali nella regolamentazione e nell'approvazione dei farmaci
L'Agenzia europea per i medicinali (EMA) ha un ruolo importante sia nella regolamentazione che nell'approvazione dei farmaci.
L'Agenzia europea per i medicinali si basa sui dati degli studi clinici svolti da aziende farmaceutiche per raggiungere le sue opinione sull'autorizzazione di farmaci. Inoltre gestisce un database di studi clinici condotti nell'Unione Europea https://www.clinicaltrialsregister.eu/
L'EMA supervisiona la procedura centralizzata (Centralised Procedure, CP) per l'autorizzazione all'immissione in commercio (Marketing Authorisation, MA). La maggioranza dei nuovi farmaci cerca autorizzazione mediante la CP: richiede all'azienda di presentare una singola domanda all'EMA e se il farmaco viene approvata una licenza viene concessa dal CE perché sia immesso in commercio in tutti i paesi dell'UE e dello SEE.
Il comitato dell'EMA responsabile per la valutazione dei dossier (domande) è il Comitato per i prodotti medicinali per uso umano (Committee for Medicinal Products for Human Use, CMHP). Gli esperti che valutano le presentazioni vengono nominati da ciascuno degli Stati membri e in più da Islanda e Norvegia. Possono essere supportati da un numero massimo di cinque membri cooptati, scelti tra esperti nominati da Stati membri o dall'EMA e arruolati, quando necessario, per fornire competenze aggiuntive in una particolare area scientifica.
A2-5.01.1-V1.1