Een geneesmiddel maken. Stap 5: Niet-klinische veiligheidstesten
Inleiding
Gemiddeld duurt het ruim 12 jaar en kost het meer dan 1 miljard euro om al het benodigde onderzoeks- en ontwikkelingswerk te doen voordat een nieuw geneesmiddel beschikbaar komt om door patiënten te worden gebruikt.
Geneesmiddelenontwikkeling is een risicovolle onderneming. De meeste stoffen (ongeveer 98%) die worden ontwikkeld, halen de markt niet als nieuw geneesmiddel. Dit komt voornamelijk doordat de voordelen en risico’s (negatieve bijwerkingen) die tijdens de ontwikkeling worden geconstateerd, zich slecht verhouden tot geneesmiddelen die al verkrijgbaar zijn voor patiënten.
De ontwikkeling van een nieuw geneesmiddel kan worden onderverdeeld in 10 verschillende stappen. Het volgende artikel gaat over Stap 5: Niet-klinische veiligheidstesten.
-
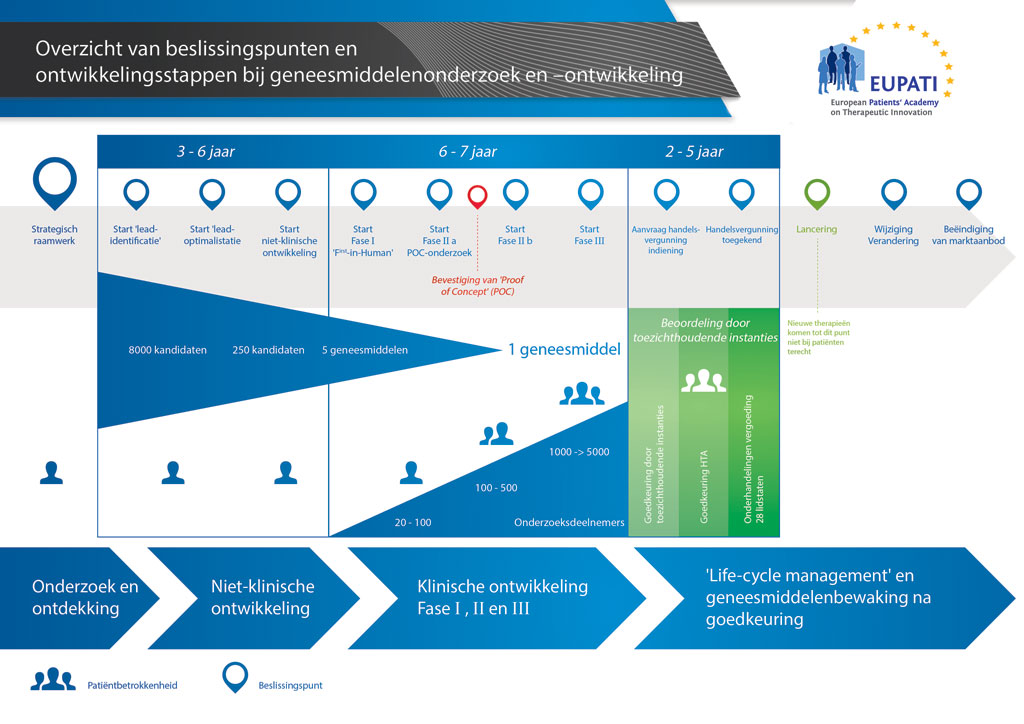
- Er kunnen meer dan 10 jaar aan nauwkeurig plannen en onderzoek nodig zijn om een geneesmiddel te ontwikkelen van een molecuul tot een verkoopbare behandeling.
Stap 5: Niet-klinische veiligheidstesten
Is het veilig om door te gaan naar de fase van klinisch testen? In dit stadium van het proces van geneesmiddelontwikkeling worden veiligheidsproeven met dieren gedaan wat wordt gereguleerd via specifieke regels en voorschriften van goede laboratoriumpraktijken (Good Laboratory Practice, GLP). Geen enkel kandidaat-geneesmiddel kan worden getest op mensen (in klinische onderzoeken) voordat het veiligheidsprofiel ervan is vastgesteld in veiligheidsproeven met dieren. Geneesmiddelenontwikkeling wordt strak gecontroleerd. De wet legt regels en voorschriften op met betrekking tot wat er wordt uitgevoerd, en hoe dat wordt uitgevoerd.
Voordat het niet-klinische testwerk kan worden gedaan, moeten grotere hoeveelheden van de kandidaat-stof worden gemaakt zodat alle proeven die van toepassing zijn, kunnen worden uitgevoerd. Dit productieproces moet ook verlopen volgens strikte richtlijnen en voorschriften, de zogeheten goede manier van produceren (Good Manufacturing Practice, GMP).
In deze voorschriften staat welke studies er moeten worden gedaan en welk type dieren moet worden gebruikt om aanvaardbare informatie te verkrijgen. Daarbij wordt onder meer gekeken naar effecten:
- bij het dier in zijn totaliteit
- in alle weefsels en organen van het dier (systemische toxiciteitsonderzoeken)
- op het voortplantingsvermogen en de normale ontwikkeling van de dieren (reproductietoxiciteitsonderzoeken)
- op de huid of ogen (lokale toxiciteitsonderzoeken)
- allergieën (overgevoeligheidsonderzoeken)
- op de chromosomen en genen (genotoxiciteitsstudies)
- effecten op kankerverwekking (carcinogeniteitsonderzoeken)
Deze studies worden hieronder getoond.
Typen toxiciteitsonderzoeken
- Systemische toxiciteitsonderzoeken
- Onderzoeken met enkelvoudige doses
- Onderzoeken met herhaalde doses
- Reproductietoxiciteitsonderzoeken
- Vruchtbaarheidsonderzoeken bij mannen
- Ontwikkelings- en reproductietoxiciteitsonderzoeken bij vrouwen
- Specifieke toxiciteitsonderzoeken
- Overgevoeligheidsonderzoeken
- Genotoxiciteitsonderzoeken
- Carcinogeniteitsonderzoeken
Deze onderzoeken tonen niet alleen het veiligheidsprofiel bij dieren, maar leveren ook belangrijke informatie op over:
- hoe de stof het lichaam binnenkomt (Absorptie)
- Distributie van de stof in het lichaam
- afbraak van de stof in het lichaam (Metabolisme)
- hoe de stof het lichaam verlaat (Excretie).
Hier wordt soms de afkorting ‘ADME’ voor gebruikt.
Al deze informatie wordt gebruikt om te beslissen of de kandidaat-stof kan doorschuiven naar het eerste bij de mens uit te voeren (klinische) onderzoek en zo ja, welke doses moeten worden gebruikt.
Om door te mogen gaan naar klinische proeven met mensen moet de kandidaat-stof een aanvaardbaar veiligheidsprofiel hebben laten zien in alle vereiste niet-klinische toxiciteitsonderzoeken. Niet alle niet-klinische veiligheidsproeven zullen dan al zijn afgerond. Langlopende carcinogeniteitsonderzoeken duren bijvoorbeeld gemiddeld twee jaar en lopen tegelijk met de klinische onderzoeken door.
Referenties
- Edwards, L., Fox, A., & Stonier, P. (Eds.). (2010). Principles and practice of pharmaceutical medicine (3rd ed.). Oxford, UK: Wiley-Blackwell.
Bijlagen
- Factsheet: Geneesmiddelontdekking
Size: 767,640 bytes, Format: .docx
Geneesmiddelontdekking. Dit feitenoverzicht bevat de stappen in het proces van geneesmiddelenontdekking en -ontwikkeling die plaatsvinden voordat een stof kan worden getest bij mensen – van ‘pre-discovery’ (informatie over de ziekte verzamelen) tot niet-klinische veiligheidsproeven met dieren.
- Presentatie: De basisprincipes van geneesmiddelontdekking en -ontwikkeling
Size: 950,426 bytes, Format: .pptx
De basisprincipes van geneesmiddelontdekking en -ontwikkeling. Gemiddeld duurt het ruim 12 jaar en kost het meer dan 1 miljard euro om al het benodigde onderzoeks- en ontwikkelingswerk te doen voordat een nieuw geneesmiddel beschikbaar komt om door patiënten te worden gebruikt. Deze presentatie gaat nader in op het proces van ontdekking tot marktintroductie van een nieuw geneesmiddel en daarna.
SA2-1.02.4-v1.1