Développement non clinique : principes de base
Introduction
La phase de développement non clinique (ou pré-clinique) vise principalement à identifier le traitement potentiel présentant la plus forte probabilité de réussite, à en évaluer la sécurité et à établir des bases scientifiques solides avant de passer à la phase de développement clinique.
En outre, pendant la phase de développement non clinique, le composé potentiel doit répondre à des objectifs non cliniques, notamment la définition de droits de propriété intellectuelle et la possibilité d’être fabriqué en quantité suffisante pour les essais cliniques. Le développement non clinique d’un médicament est complexe et réglementé.
Notions élémentaires, définitions et concepts
Non clinique ou pré-clinique ?
Les termes « non clinique » et « pré-clinique » sont souvent utilisés comme synonymes.
Bien que leur importance soit cruciale en phase pré-clinique du développement, les études non cliniques peuvent être effectuées à tout moment du cycle de vie du produit, le mieux étant aussi tôt que possible afin d’éviter les surprises ultérieures pendant le développement.
Au-delà de l’identification des propriétés pharmacodynamiques (l’effet d’un médicament sur le corps), pharmacocinétiques (l’effet du corps sur le médicament) et toxicologiques du composé étudié avant administration à des sujets humains, les données des études non cliniques permettent d’affiner, de consolider et de compléter les informations ; le profil de sécurité du produit peut ainsi être mis à jour au cours de la phase pré-clinique, lors de l’enregistrement et tout au long du cycle de vie du médicament.
Études in silico, in vitro et in vivo
Les études non cliniques sont effectuées :
- In silico : sur ordinateur ou par simulation logicielle, par ex. prévision du profil toxicologique d’un produit à partir de sa structure chimique identifiée dans des bases de données.
- In vitro (locution latine signifiant « dans le verre ») : étude effectuée en environnement contrôlé, à l’extérieur d’un organisme vivant, par ex. utilisation de cultures d’hépatocytes (cellules du foie) pour des études de métabolisme.
- In vivo (locution latine signifiant « dans le vivant ») : expérimentation sur un organisme vivant entier, par opposition à des tissus ou des cellules, c’est-à-dire des animaux, des humains ou des plantes.
Quels sont les aspects essentiels de chimie, fabrication et contrôle (CMC) pendant le développement non clinique ?
Toutes les études de développement non clinique nécessitent la fabrication d’un principe actif adéquat :
- De faibles quantités (de l’ordre du milligramme ou du gramme) sont généralement nécessaires pour les études non cliniques ; un accroissement de l’échelle de production est ensuite nécessaire pour fournir les quantités suffisantes aux essais cliniques, puis, après obtention de l’autorisation, les quantités exigées par le marché.
- Pour des études conformes aux bonnes pratiques de laboratoire (BPL), des lots de principe actif qualifiés ou conformes aux bonnes pratiques de fabrication (BPF) doivent être fournis.
Voici quelques-unes des étapes CMC essentielles pendant la phase de développement non clinique :
- Détermination de la posologie et du mode d’administration
- Caractérisation physico-chimique détaillée
- Tests de stabilité et analyse des impuretés
- Développement et validation des méthodes analytiques pour quantifier le principe actif dans les liquides corporels comme le sang, le plasma, l’urine lors des études d’activité et d’effets secondaires
- Développement d’un prototype pour ce qui sera utilisé en clinique.
Processus de développement non clinique
Activités de développement non clinique parallèles aux activités de recherche. Elles doivent supporter le programme de développement clinique planifié en répondant aux objectifs et questions décrits ci-dessous.
Objectifs
Lorsqu’un composé potentiel est identifié, le développement non clinique doit commencer à répondre aux questions suivantes et les réponses doivent être fournies par des évaluations/études spécifiques :
- Le composé est-il actif ? → évaluation de l’efficacité
- Comment sera-t-il administré et comment réagira le corps ? → étude du profil
- Est-il sans danger ? → toxicologie/tolérance
- La fabrication est-elle viable et contrôlable ?
Les activités de développement non clinique peuvent continuer pendant tout le cycle de vie du produit ; toutefois, plus ces questions sont résolues rapidement, plus il est facile d’identifier le profil de patient qui bénéficiera le plus du composé.
Gestion de projet
Le programme de développement non clinique est complexe et nécessite des compétences solides en gestion de projet et en communication pour encadrer des équipes pluridisciplinaires. L’équipe du projet doit appréhender le plan clinique afin de définir le programme non clinique et les activités associées.
Le profil fournit un cadre pour la stratégie de développement non clinique, définissant des objectifs, le risque, des responsabilités, des indicateurs et un processus de décision Go/No-go (Poursuite/Arrêt du projet). La mise en œuvre du profil permet plus facilement d’axer le projet sur les principaux critères du produit, de prendre des décisions de poursuite (Go/No go) en temps voulu et de réduire le risque global du projet (poursuivre le développement d’un produit sans intérêt).
Recommandations réglementaires non cliniques
De nombreux acteurs sont impliqués dans le développement de médicaments et chaque organisme ou institution suit ses propres règles. Les entreprises ont par exemple leurs propres procédures opérationnelles standard (POS). Le site Internet de l’Agence européenne des médicaments (EMA) fournit les Bonnes pratiques cliniques, ainsi que des recommandations.
- Elles sont générales ou plus spécifiques et traitent des aspects scientifiques et techniques (par ex. spécifiques aux études toxicologiques requises).
- Elles doivent être strictement respectées pour toute demande d’autorisation de mise sur le marché ; tout écart doit être justifié.
Le dossier de demande doit être soumis au format DTC (Document technique commun) qui a été défini par la Conférence internationale sur l’harmonisation des exigences techniques pour l’enregistrement des médicaments à usage humain (ICH). La décision de rassembler toutes les informations de qualité, de sécurité et d’efficacité dans un format commun (le DTC) a révolutionné le processus d’examen réglementaire et permet une soumission électronique harmonisée et la mise en œuvre de bonnes pratiques d’examen. Le DTC évite aux industriels de devoir reformater les dossiers de demande pour chaque autorité réglementaire (l’ICH rassemble les autorités réglementaires et l’industrie pharmaceutique de l’Union européenne, du Japon et des États-Unis pour discuter des aspects scientifiques et techniques de l’enregistrement des médicaments).
-
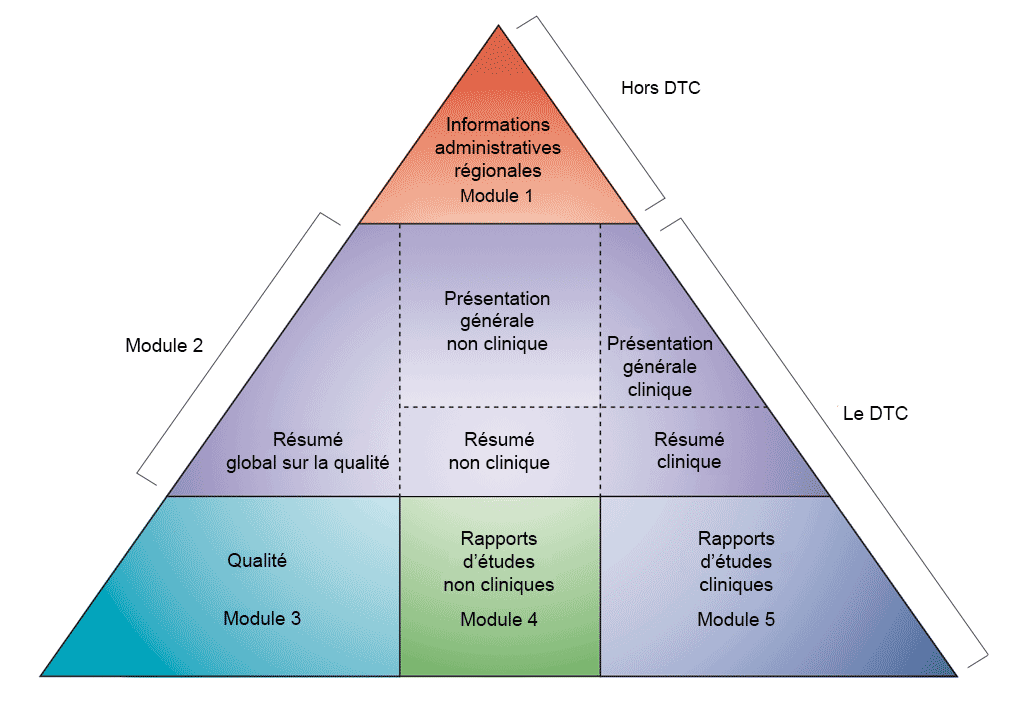
- Développement non clinique dans des modules CTD. Adaptation du CTD ICH (voir référence 1)
Le DTC comporte 5 modules différents (voir la figure ci-dessus). En juillet 2003, le DTC est devenu le format obligatoire pour toute nouvelle demande d'autorisation de mise sur le marché dans l'Union européenne et au Japon ; aux États-Unis, ce format est fortement recommandé pour toute demande d'autorisation de nouveau médicament (New Drug Application, NDA) soumise à la FDA (Food and Drug Administration).
Résumé
La phase de développement non clinique est cruciale et doit anticiper les problèmes potentiels avant que le composé ne passe en phase de développement clinique.
L'admission d'un composé potentiel en phase d'étude clinique requiert les éléments suivants :
- Évaluation de sécurité non clinique obtenue dans le respect des Bonnes pratiques de laboratoire (BPL).
- Fabrication soumise à un contrôle qualité rigoureux.
- Données et processus présentés au format DTC, fournissant une base solide pour la phase de développement clinique.
On observe une tendance croissante à concevoir des propriétés de type médicamenteux in silico, ainsi qu'à utiliser des méthodes bio-informatiques dans la modélisation et la prévision.
Les recommandations de développement non clinique font l'objet d'une harmonisation continue entre les principales autorités réglementaires (Europe, États-Unis et Japon) qui porte en priorité sur la sécurité et la qualité : l'ICH fournit régulièrement des recommandations détaillées pour l'industrie pharmaceutique, similaires à celles publiées par les agences européenne (EMA) et américaine (FDA).
Ressources complémentaires
- European Medicines Agency (2007a). Guideline on strategies to identify and mitigate risks for first-in human trials with investigational medicinal products. London: European Medicines Agency. Retrieved 25 June, 2015, from http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002988.pdf
- European Medicines Agency (2007b). Guideline on requirements for first-in-man clinical trials for potential high-risk medicinal Retrieved 25 June, 2015, from http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002989.pdf
- European Medicines Agency (2009). ICH guideline M3(R2) on non-clinical safety studies for the conduct of human clinical trials and marketing authorisation for pharmaceuticals. London: European Medicines Agency. Retrieved 25 June, 2015, from http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002720.pdf
- European Medicines Agency (2011). Committee for medicinal products for human use (CHMP) ICH guidelines S6 (R1) – preclinical safety evaluation of biotechnology-derived pharmaceuticals. London: European Medicines Agency. Retrieved 25 June, 2015, from http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002828.pdf
[glossary_exclude]Références
- Image reproduced from ICH (2015). M4: The Common Technical Document. Retrieved 11 July, 2021, from https://www.ich.org/page/ctd
- European Medicines Agency (2009). ICH guideline M3(R2) on non-clinical safety studies for the conduct of human clinical trials and marketing authorisation for pharmaceuticals. London: European Medicines Agency. Retrieved 25 June, 2015, from http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002720.pdf[/glossary_exclude]