Der prädiktive Wert präklinischer Prüfungen
Einleitung
Beginnend mit den ersten Schritten des Arzneimittelentdeckungs- und -entwicklungsprozesses sind die in präklinischen Untersuchungen ermittelten Daten für die Entscheidungsfindung hinsichtlich Wirksamkeit und Sicherheit unverzichtbar – so beispielsweise für die Risikomanagement-Planung, die Risikominderung, die Bedingungen und Spezifikationen der Marktzulassung, die Anwendung des Arzneimittels auf dem Markt und die Sicherheitsüberwachung nach Markteinführung (Pharmakovigilanz).
Im Rahmen präklinischer Untersuchungen gewonnene Informationen spielen eine Schlüsselrolle bei Entscheidungen zu:
- klinischen Studien
- Risikomanagement und -minderung
- Antrag auf Marktzulassung
- Verschreibung eines Arzneimittels für einen Patienten
- Post-Markteinführungs- und Marktüberwachungsstudien
- und weiteres
Das folgende Rahmenkonzept illustriert im Arzneimittelentdeckungs- und -entwicklungsprozess entscheidende Bedürfnisse und Faktoren. Präklinische Informationen spielen eine wichtige Rolle bei der Bestimmung dieser Bedürfnisse und Faktoren. Dieser Artikel erforscht die wichtige Rolle präklinischer Studien als bedeutenden Prädiktor für klinische Studien am Menschen.
Die Schlüssel für eine erfolgreiche Arzneimittelentwicklung1
- Richtiges „Target“
- Enge Korrelation zwischen dem „Target“ des Arzneimittels und der Erkrankung
- Verfügbare und prädiktive Biomarker
- Richtiges Gewebe
- Adäquate Bioverfügbarkeit und Gewebe-Exposition
- Definition von Pharmakodynamik-Biomarkern
- Eindeutiges Verständnis der präklinischen und klinischen Pharmakokinetik und Pharmakodynamik
- Verständnis der Wechselwirkungen mit anderen Arzneimitteln (Wirkstoff-Wirkstoff-Wechselwirkungen)
- Richtige Sicherheit
- Eindeutige Sicherheitsmargen
- Verständnis der sekundären pharmakologischen Risiken
- Verständnis der reaktiven Metaboliten, der Genotoxizität und der Wechselwirkungen mit anderen Arzneimitteln
- Verständnis der gefährlichen Nebenwirkungen und anderer Anfälligkeiten
- Richtige Patienten
- Identifizierung der am besten auf die Behandlung ansprechenden Patientenpopulation
- Definition des Nutzen-Risiko-Verhältnisses für die jeweilige Population
- Richtiges wirtschaftliches Potenzial
- Kosten/Nutzen vs. zukünftiger Versorgungsstandard
- Fokus auf Marktzugang
Von Laborstudien und Studien am Tiermodell zur Behandlung von Patienten
Ein Wirkstoffkandidat kann erst dann an Menschen verabreicht werden, wenn in ausreichendem Maß unterstützende Informationen hinsichtlich seines Sicherheitsprofils und der erwarteten Wirkungen zusammengetragen wurden. Diese unterstützenden Informationen werden im Rahmen präklinischer Studien gewonnen, die wichtige Prädiktoren wie den „Machbarkeitsnachweis“, Dosierungsvorschläge, angemessene Sicherheitsüberwachung und geeignete Einschluss- und Ausschlusskriterien liefern.
Präklinische Untersuchungen am Zell- (in vitro) und Tiermodell (in vivo) sollten daher:
- die Wirksamkeit des Wirkstoffkandidaten nachweisen
- Erkenntnisse zum Sicherheitsprofil des Wirkstoffkandidaten beschaffen (beispielsweise in Untersuchungen zur maximal verträglichen Dosis)
- Auswirkungen des Wirkstoffkandidaten abschätzen, die am Menschen nicht erforscht werden können – beispielsweise Auswirkungen der Verbindung auf Schwangere oder die Leibesfrucht
Extrapolation vom Tiermodell auf den Menschen
Die Extrapolation von in Laboruntersuchungen und im Tiermodell gewonnenen Informationen zu einem Arzneimittel auf die Anwendung beim Menschen erfordert professionellen Sachverstand. In den Leitlinien des Ausschusses für Arzneimittel für die Anwendung am Menschen (CHMP – Committee for Human Medicinal Products)2 der Europäischen Arzneimittel-Agentur sowie der Internationalen Harmonisierungskonferenz (International Conference on Harmonisation, ICH)3 finden sich nützliche Regeln, die für derartige Extrapolationsprozesse entwickelt wurden. Diese Leitlinien legen die Art der vor Durchführung klinischer Studien zu absolvierenden Untersuchungen fest.
Im präklinischen Programm zu einem Wirkstoffkandidaten begründete Probleme können Ursachen für Einwände bei der Evaluierung des Antrags auf Marktzulassung (Marketing Authorisation Application, MAA) im Rahmen des behördlichen Zulassungsprozesses sein. Derartige Fälle führen zu Fragen hinsichtlich der Relevanz der für die vorgesehene Indikation, d. h. die mit dem Wirkstoffkandidaten zu behandelnde Erkrankung, des Wirkstoffkandidaten verwendeten präklinischen Modells. Um derartige Probleme zu vermeiden, müssen präklinische Studien sorgfältig geplant werden, damit die in Labor- und Tierstudien generierten Erwartungen als zufriedenstellende Prädiktoren fungieren können.
Das Ausmaß und der Umfang des präklinischen Programms, das vor der Aufnahme klinischer Studien zufriedenstellend abgeschlossen werden muss, bestimmen sich nach den folgenden Faktoren:
- die Art und der Schweregrad der zu behandelnden Erkrankung
- die Größe und Dynamik der Patientenpopulation, für deren Behandlung der Wirkstoffkandidat vorgesehen ist
- die Phase der klinischen Studie (Phase I, II, III oder IV [Sicherheitsüberwachung nach Markteinführung])
- die voraussichtliche Dosis und Dauer der Behandlung beim Menschen
Diese Erwägungen dienen zur Rechtfertigung der Art der Untersuchungen bzw. der im Rahmen des präklinischen Programms eingesetzten Tiermodelle.
Viele Unternehmen lassen sich von den Aufsichtsbehörden (z. B. Europäische Arzneimittel-Agentur [EMA] oder nationale Gesundheitsbehörde) hinsichtlich präklinischer Studien wissenschaftlich beraten. Diese wissenschaftliche Beratung unterstützt das Unternehmen dabei, sicherzustellen, dass die geeigneten Tests und Untersuchungen durchgeführt werden, so dass im Rahmen des Antrags auf Marktzulassung wesentliche Einwände hinsichtlich des Designs der Tests und der Untersuchungen eher nicht zu erwarten sind. Die Einholung und Befolgung des Rats dieser Stellen erhöht die Wahrscheinlichkeit eines positiven Ergebnisses im MAA-Stadium (Antrag auf Marktzulassung). Diese Beratung erfolgt im Licht des aktuellen wissenschaftlichen Kenntnisstands und basiert auf der vom Unternehmen bereitgestellten Dokumentation.
Präklinische Daten besitzen in den frühen Abschnitten des Prozesses der Entwicklung eines Wirkstoffkandidaten die größte Bedeutung (siehe Abbildung 1). Bis ein Arzneimittel für die Verschreibung an Patienten zur Verfügung steht (nach Erteilung der Marktzulassung), wurde ein Großteil der präklinischen Daten zur Sicherheit und Wirksamkeit durch Daten aus klinischen Studien am Menschen ersetzt. In manchen Fällen jedoch – beispielsweise bei der Bestimmung der Auswirkungen eines Wirkstoffkandidaten auf die Entwicklung von Krebs oder auf das Reproduktionsvermögen – verhindern ethische Bedenken die Gewinnung von Daten am Menschen. In Fällen wie diesen richtet sich die klinische Anwendung neuer Arzneimittel über einen längeren Zeitraum nach den aus präklinischen Daten gewonnenen Erkenntnissen. Letztlich jedoch müssen auch diese durch in derartigen Fällen (z. B. im Rahmen des Lebenszyklus-Managements nach Marktzulassung oder der Pharmakovigilanz) gewonnene Daten ersetzt werden.
-
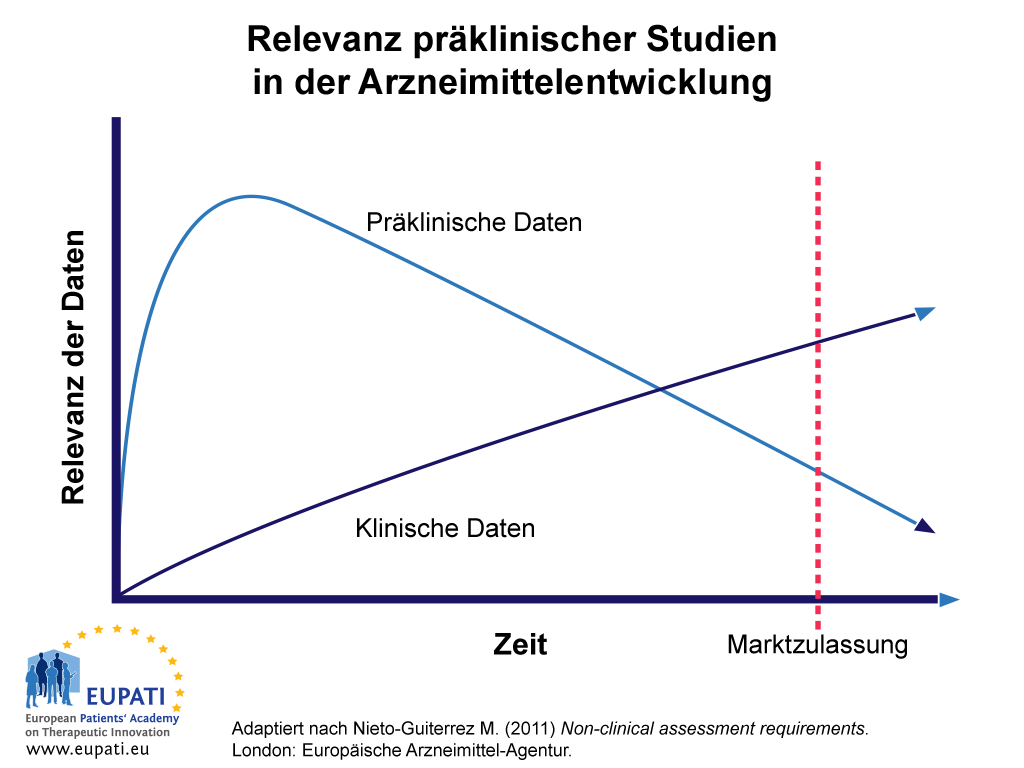
- Während präklinische Daten zu Beginn der Entwicklungsphase eine wesentlich höhere Relevanz besitzen, wird diese im Laufe der Zeit von der klinischer Daten überholt.
Abbildung 1 veranschaulicht die relative Bedeutung und Zuverlässigkeit präklinischer Daten im Arzneimittelentwicklungsprozess im Verlauf der Zeit. Daten aus präklinischen Studien erfahren bis zu einem späteren Zeitpunkt im Entwicklungsprozess größeres Vertrauen als klinische Daten.
Idealerweise sollten alle in der Entwicklungsphase aufgeworfenen präklinischen Sicherheitsbedenken behoben sein, wenn der Antrag auf Marktzulassung gestellt wird. Tatsächlich können jedoch zum Zeitpunkt der Einreichung und Beurteilung des Dossiers wesentliche Ursachen für Sicherheitsbedenken wie beispielsweise Karzinogenität, Genotoxizität, genotoxische Verunreinigungen, Reproduktionstoxizität und Hepatotoxizität weiterhin vorliegen.
Ethische Betrachtungen
Die Zulässigkeit der Verwendung von Tieren als Modell für die Beurteilung des Risikos beim Menschen und der Verwendung derartiger Modelle für die Nachahmung von Erkrankungen des Menschen ist in der Erklärung von Helsinki festgeschrieben.4 Die Erklärung von Helsinki beinhaltet auch die ethische und wissenschaftliche Rechtfertigung für die erste Exposition des Wirkstoffkandidaten gegenüber gesunden Freiwilligen. Darüber hinaus konstatiert die Erklärung, dass biomedizinische Forschung auf adäquat durchgeführten Labor- und Tierexperimenten und gründlicher Kenntnis der wissenschaftlichen Literatur fußen sollte. Das Wohlergehen der für Forschungszwecke eingesetzten Tiere ist zu respektieren.
Präklinische Studien: Angemessene Prädiktoren für Untersuchungen am Menschen?
In der Vergangenheit standen die Herausforderungen für den prädiktiven Wert präklinischer Studien im Zusammenhang mit Aspekten der Pharmakokinetik, der Pharmakodynamik (Wirksamkeit) und der Sicherheit beim Menschen, die sich der Prädiktion durch präklinische Studien leicht entziehen. Viele neue Technologien (in silico [Computermodelle], Pharmakogenomik, Biomarker, neuartige explorative Designs für klinische Studien usw.), die sämtlich einen positiven Einfluss auf den prädiktiven Wert präklinischer Studien aufweisen, befinden sich in rasanter Entwicklung.
Weitergehende Informationen
- Die Erklärung von Helsinki steht unter Retrieved 13 July, 2021, from https://www.wma.net/policies-post/wma-declaration-of-helsinki-ethical-principles-for-medical-research-involving-human-subjects/ (Stand: 23. September 2015) in den Sprachen Englisch, Spanisch und Französisch zur Verfügung. Unter http://www.wma.net/en/20activities/10ethics/10helsinki/ (Stand: 23. September 2015) ist sie auch in den Sprachen Deutsch, Japanisch, Portugiesisch, Tschechisch und Ungarisch verfügbar.
Quellenangaben
- Adapted from Cook, D., Brown, D., Alexander, R., March, R., Morgan, P., Satterthwaite, G., & Pangalos, M. (2014). Lessons learned from the fate of AstraZeneca's drug pipeline: A five-dimensional framework. Nature Reviews Drug Discovery, 13, 419-431. doi:10.1038/nrd4309
- European Medicines Agency. (2015) Non-clinical guidelines. Retrieved 24 July, 2015, from http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_000083.jsp&mid=WC0b01ac0580027548
- International Conference on Harmonisation (2015). ICH Guidelines. Retrieved 24 July, 2015, from http://www.ich.org/products/guidelines.html
- World Medical Association. (2013) WMA Declaration of Helsinki – Ethical Principles for Medical Research Involving Human Subjects. Retrieved 24 July, 2015, from http://www.wma.net/en/30publications/10policies/b3/
- Nieto-Guiterrez, M. (2011) Non-clinical Assessment Requirements. Brussels: European Medicines Agency. Retrieved 24 July, 2015, from http://www.ema.europa.eu/docs/en_GB/document_library/Presentation/2011/06/WC500107868.pdf
Anlagen
- Präsentation: Nicht-klinische Entwicklung
Size: 396,736 bytes, Format: .pptx
Eine Präsentation zum nicht-klinischen Entwicklungsabschnitt eines Arzneimittels. Diese Präsentation behandelt Ziele der nicht-klinischen Entwicklung, Hintergrundaktivitäten (einschließlich der Herstellung des benötigten Wirkstoffs), Arten von nicht-klinischen Studien, spezifische Besonderheiten des Tiermodells, Abwägungen zum zeitlichen Ablauf und der Dauer sowie die nicht-klinischen Ergebnisse, die zu einer Einstellung der Entwicklung eines Wirkstoffkandidaten führen können.
A2-2.02.1-v1.3