De voorspellende waarde van niet-klinisch testen
Inleiding
Vanaf het begin van het proces van ontdekking en ontwikkeling van geneesmiddelen zijn gegevens die zijn verzameld in niet-klinische onderzoeken essentieel voor besluitvorming inzake werkzaamheid en veiligheid – bijvoorbeeld betreffende risicomanagementplanning, risicovermindering, de voorwaarden en specificaties van handsvergunningen, het gebruik van het geneesmiddel op de markt en veiligheidsmonitoring (farmacovigilantie) na verlening van de handelsvergunning (postmarketing).
De informatie die wordt verzameld tijdens niet-klinische onderzoeken, speelt een sleutelrol bij beslissingen inzake:
- klinische onderzoeken;
- risicomanagement en -vermindering
- aanvragen van handelsvergunningen;
- het voorschrijven van een geneesmiddel aan een patiënt;
- postmarketing- of monitoringsonderzoeken
- en dergelijke.
Het onderstaande raamwerk illustreert de behoeften en factoren die cruciaal zijn tijdens het proces van ontdekking en ontwikkeling van geneesmiddelen. Niet-klinische informatie speelt een sleutelrol bij het bepalen van deze behoeften en factoren. Dit artikel onderzoekt de belangrijke rol van niet-klinische onderzoeken als een belangrijke voorspellende factor voor klinische onderzoeken bij patiënten.
De sleutels voor de succesvolle ontwikkeling van geneesmiddelen – het ‘raamwerk van de vijf J’s’1
- Juiste doel
- Sterke correlatie tussen het doel van het geneesmiddel en de ziekte
- Beschikbare en voorspellende biomarkers
- Juiste weefsel
- Toereikende biologische beschikbaarheid en blootstelling van weefsel
- Definitie van farmacodynamische biomarkers
- Goed inzicht in de preklinische en klinische farmacokinetiek en farmacodynamie
- Inzicht in de interacties met andere geneesmiddelen (geneesmiddelinteracties)
- Juiste veiligheid
- Duidelijk veiligheidsgrenzen
- Inzicht in secundaire farmacologische risico’s
- Kennis van reactieve metabolieten, genotoxiciteit en interacties met andere geneesmiddelen
- Kennis van gevaarlijke bijwerkingen en andere risico’s
- Juiste patiënten
- Identificatie van de patiëntenpopulatie met de beste respons
- Definitie van de relatie tussen voordelen en risico’s voor de betreffende populatie
- Juiste commerciële potentie
- Kosten-baten versus toekomstige zorgstandaard
- Focus op toegang tot de markt
Van laboratorium- en dieronderzoek naar patiënten
Een kandidaat-verbinding kan niet aan patiënten worden toegediend voordat er voldoende ondersteunende informatie is verzameld over het veiligheidsprofiel van het middel en de verwachte effecten. Niet-klinische onderzoeken verschaffen deze ondersteunende informatie door te voorzien in belangrijke voorspellende factoren, zoals ‘proof of concept’, een voorgesteld doseringsschema, adequate veiligheidsmonitoring en de juiste inclusie- en exclusiecriteria.
Niet-klinische onderzoeken in cellen (in vitro) en dieren (in vivo) moeten daarom:
- de werkzaamheid van de kandidaat-verbinding aantonen,
- kennis opleveren over het veiligheidsprofiel van de kandidaat-verbinding, bijvoorbeeld onderzoeken die de maximale verdraagbare dosis beoordelen en
- de effecten schatten van de kandidaat-verbinding die niet bij de mens kunnen worden onderzocht – bijvoorbeeld het effect van de verbinding op een foetus en bij zwangere vrouwen.
Extrapoleren van dieren naar mensen
Extrapolatie van in het laboratorium en bij dieronderzoek verzamelde informatie naar toepassing van een geneesmiddel bij mensen vereist een professioneel oordeel. In de richtlijnen van het Comité voor geneesmiddelen voor menselijk gebruik (CHMP) van het Europees Geneesmiddelenbureau (EMA) en de International Conference on Harmonisation (ICH) zijn2 bruikbare regels voor de extrapolatieprocedures ontwikkeld en beschreven3. Deze richtlijnen specificeren de typen onderzoeken die moeten plaatsvinden voordat er klinische onderzoeken kunnen worden uitgevoerd.
Problemen met het niet-klinische programma van de kandidaat-verbinding kunnen bezwaren oproepen tijdens de evaluatie van de aanvraag van de handelsvergunning tijdens de beoordeling door toezichthoudende instanties. Dergelijke gevallen leiden tot vragen over de relevantie van de niet-klinische modellen die zijn gebruikt voor de voorgestelde indicatie, nl. dat de kandidaat-verbinding bedoeld is voor de behandeling van mensen. Om deze problemen te vermijden moeten niet-klinische onderzoeken zorgvuldig worden gepland zodat de verwachtingen gewekt door laboratorium- en dieronderzoek, als goede voorspellende factoren kunnen fungeren.
De omvang en reikwijdte van het niet-klinische programma dat op bevredigende wijze moet zijn afgerond voordat er klinische onderzoeken kunnen worden gestart, variëren afhankelijk van de volgende factoren:
- het type en de ernst van de beoogde ziekte;
- de omvang en dynamiek van de populatie waarvoor de kandidaat-verbinding als behandeling bedoeld is;
- de klinische onderzoeksfase (fase I, II, III en fase IV na vergunningverlening) en
- de verwachte dosis en duur van de behandeling bij mensen.
Deze overwegingen worden gebruikt voor onderbouwing van de soorten tests of diermodellen die tijdens het niet-klinische programma worden gebruikt.
Veel bedrijven winnen bij toezichthoudende instanties (bijvoorbeeld het Europees Geneesmiddelenbureau (EMA) of nationale bevoegde autoriteiten) wetenschappelijk advies in over niet-klinische onderzoeken. Dit wetenschappelijke advies draagt ertoe bij dat het bedrijf de juiste tests en onderzoeken uitvoert, zodat de kans afneemt dat er tijdens de aanvraag van de handelsvergunning ernstige bezwaren inzake de opzet van de tests naar voren komen. Door bij deze bureaus advies in te winnen en dit op te volgen neemt de kans op een positief resultaat wat betreft de aanvraag van een handelsvergunning toe. Het advies wordt gegeven in het licht van de actuele wetenschappelijke kennis en is gebaseerd op de door het bedrijf overgelegde documentatie.
Niet-klinische gegevens zijn het belangrijkst in de vroege fasen van het ontwikkelingsproces van een kandidaat-verbinding (zie afbeelding 1). Tegen de tijd dat een geneesmiddel kan worden voorgeschreven (na verlening van de handelsvergunning), is een groot gedeelte van de niet-klinische gegevens over veiligheid en werkzaamheid vervangen door gegevens afkomstig van klinische onderzoeken bij patiënten. In sommige gevallen echter – bijvoorbeeld het effect van een kandidaat-verbinding op de ontwikkeling van kanker of op de voortplanting – verhinderen ethische overwegingen het verzamelen van gegevens bij proefpersonen. In deze gevallen vindt het klinische gebruik van nieuwe geneesmiddelen gedurende een langere periode plaats op geleide van niet-klinische gegevens. Uiteindelijk moeten deze echter ook worden vervangen door later verzamelde gegevens, bijvoorbeeld als onderdeel van ‘life-cycle management’ en geneesmiddelenbewaking na vergunningverlening.
#mla_gallery-1 { margin: auto; width: 100%; } #mla_gallery-1 .gallery-item { float: none; margin: 1.5%; display: inline-block; text-align: center; width: 97%; } #mla_gallery-1 .gallery-item .gallery-icon img { border: 2px solid #cfcfcf; } #mla_gallery-1 .gallery-caption { margin-left: 0; vertical-align: top; } /* see mla_gallery_shortcode() in media-library-assistant/includes/class-mla-shortcode-support.php */
-
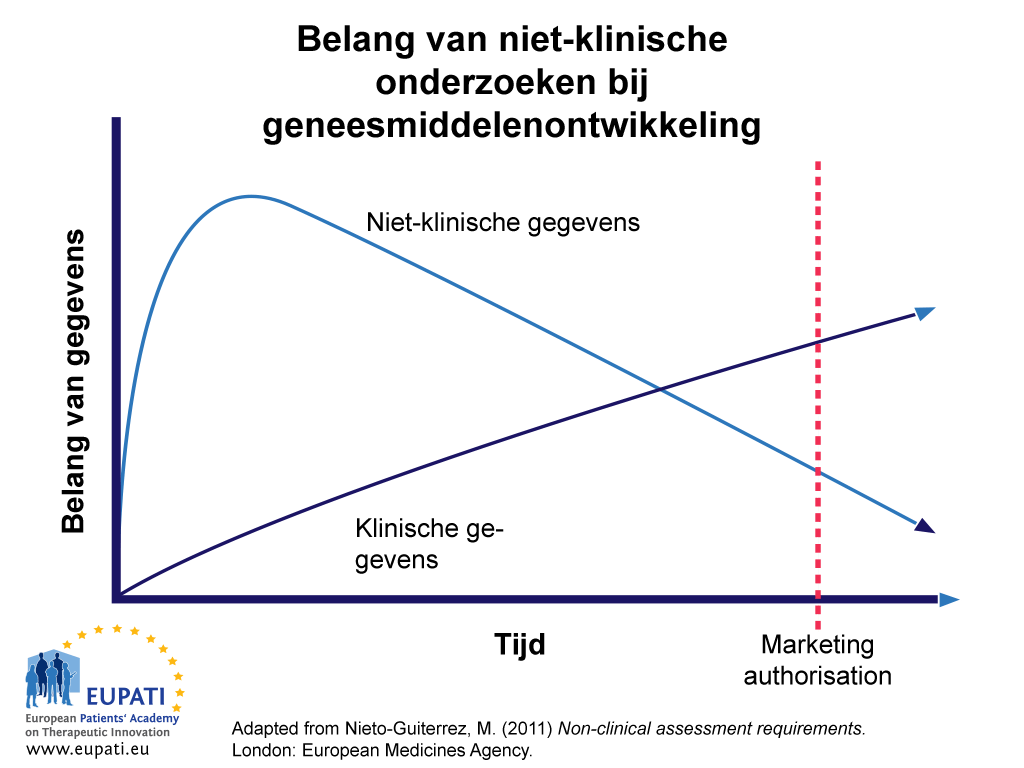
- Hoewel niet-klinische gegevens aan het begin veel belangrijker zijn voor het proces van geneesmiddelenontwikkeling, wordt hun betekenis in de loop van de tijd overschaduwd door die van klinische gegevens.
Afbeelding 1 geeft het relatieve belang van en vertrouwen op niet-klinische gegevens weer bij het proces van geneesmiddelenontwikkeling in de loop van de tijd. In eerste instantie wordt er meer vertrouwd op gegevens van niet-klinische onderzoeken dan op klinische gegevens, die pas later in het ontwikkelingsproces belangrijk worden.
In het ideale geval moeten alle niet-klinische veiligheidsproblemen die zich voordeden in de ontwikkelingsperiode, zijn opgelost tegen de tijd dat de aanvraag van de handelsvergunning wordt ingediend. Wanneer het dossier wordt ingediend en beoordeeld, kunnen er echter nog steeds grote zorgen bestaan inzake de veiligheid, zoals bijvoorbeeld carcinogeniciteit, genotoxiciteit, genotoxische onzuiverheden, reproductietoxiciteit en hepatotoxiciteit.
Ethische overwegingen
De aanvaardbaarheid van het gebruik van dieren als modellen voor risicobeoordeling bij mensen en voor het gebruik van deze modellen voor het nabootsen van menselijke ziekten wordt uiteengezet in de Verklaring van Helsinki.4 De Verklaring van Helsinki biedt de ethische en wetenschappelijk rechtvaardiging voor de eerste blootstelling van kandidaat-verbindingen bij gezonde vrijwilligers. Bovendien vermeldt de Verklaring dat biomedisch onderzoek moet worden gebaseerd op adequaat uitgevoerde laboratorium- en dierexperimenten en op grondige kennis van de wetenschappelijk literatuur. Het welzijn van dieren die voor onderzoek worden gebruikt, moet worden gerespecteerd.
Niet-klinische onderzoeken: geschikte voorspellende factoren voor onderzoeken bij mensen?
Vroeger waren problemen met de voorspellende waarde van niet-klinische onderzoeken gerelateerd aan farmacokinetiek, farmacodynamie (werkzaamheid) en veiligheidsaspecten bij mensen, die niet gemakkelijk kunnen worden voorspeld door niet-klinische onderzoeken. Veel nieuwe technologieën in silico (computermodellen), farmacogenomica, biomarkers en baanbrekende exploratieve ontwerpen voor klinische onderzoeken ontwikkelen zich snel en hebben allemaal een positieve invloed op de voorspellende waarde van niet-klinische onderzoeken.
Overige informatiebronnen
- De Verklaring van Helsinki Retrieved 13 July, 2021, from https://www.wma.net/policies-post/wma-declaration-of-helsinki-ethical-principles-for-medical-research-involving-human-subjects/ (geraadpleegd op 23 september 2015). Het document is eveneens beschikbaar in het Duits, Japans, Portugees, Tsjechisch en Hongaars op http://www.wma.net/en/20activities/10ethics/10helsinki/ (geraadpleegd op 23 september 2015)
De Verklaring van Helsinkiis beschikbaar in het Engels, Spaans en Frans op https://www.wma.net/policies-post/wma-declaration-of-helsinki-ethical-principles-for-medical-research-involving-human-subjects/ (Retrieved 4, July 2021). Het document is eveneens beschikbaar in het Tsjechisch, Duits en Portugees, op https://web.archive.org/web/20160517043747/http://www.wma.net/en/20activities/10ethics/10helsinki (Geraadpleegd 4 juli 2021)
Referenties
- Naar Cook, D., Brown, D., Alexander, R., March, R., Morgan, P., Satterthwaite, G., & Pangalos, M. (2014). Lessons learned from the fate of AstraZeneca’s drug pipeline: A five-dimensional framework. Nature Reviews Drug Discovery, 13, 419-431. doi:10.1038/nrd4309
- European Medicines Agency. (2015) Non-clinical guidelines. Geraadpleegd op 24 juli 2015 op: http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_000083.jsp&mid=WC0b01ac0580027548
- International Conference on Harmonisation (2015). ICH Guidelines. Geraadpleegd op 24 juli 2015 op: http://www.ich.org/products/guidelines.html
- World Medical Association. (2013) WMA Declaration of Helsinki – Ethical Principles for Medical Research Involving Human Subjects. Geraadpleegd op 24 juli 2015 op: http://www.wma.net/en/30publications/10policies/b3/
- Nieto-Guiterrez, M. (2011) Non-clinical Assessment Requirements. Brussels: European Medicines Agency. Geraadpleegd op 24 juli 2015 op: http://www.ema.europa.eu/docs/en_GB/document_library/Presentation/2011/06/WC500107868.pdf
Bijlagen
#mla_gallery-2 { margin: auto; width: 100%; } #mla_gallery-2 .gallery-item { float: none; margin: 1.5%; display: inline-block; text-align: center; width: 97%; } #mla_gallery-2 .gallery-item .gallery-icon img { border: 2px solid #cfcfcf; } #mla_gallery-2 .gallery-caption { margin-left: 0; vertical-align: top; } /* see mla_gallery_shortcode() in media-library-assistant/includes/class-mla-shortcode-support.php */
- Presentatie: Niet-Klinische Ontwikkeling
Size: 498,813 bytes, Format: .pptx
Presentatie over de aspecten van niet-klinische ontwikkeling, zoals doelstellingen, achtergrondactiviteiten en de verschillende vormen van niet-klinisch onderzoek.
A2-2.02.1-v1.3