Bioverfügbarkeit und Bioäquivalenz
Bioverfügbarkeit
Bioverfügbarkeit ist wie folgt definiert: Der Anteil (Prozentsatz) der verabreichten Dosis des Arzneimittels, der unmodifiziert in die Blutbahn gelangt (systemische Zirkulation).
Bei jeder Anwendung eines Arzneimittels soll der Wirkstoff des Arzneimittels (API, Active Pharmaceutical Ingredient) in den Körper gelangen können. Um eine therapeutische Wirkung zu erzielen, genügt es jedoch nicht, dass der Wirkstoff in den Körper gelangt. Der Wirkstoff muss an der konkreten Stelle im Körper, wo er wirken soll, und in der korrekten Dosierung vorliegen. Diese spezifische Stelle wird als „Wirkort“ bezeichnet. Zudem muss der Wirkstoff den Wirkort innerhalb einer gewissen Zeit erreichen und dort für eine definierte Zeit verfügbar sein.
Bei einer Injektion direkt in die Blutbahn, d. h. einer intravenösen Injektion, beträgt die Bioverfügbarkeit definitionsgemäß 100 % (siehe nachstehende Abbildung).
-
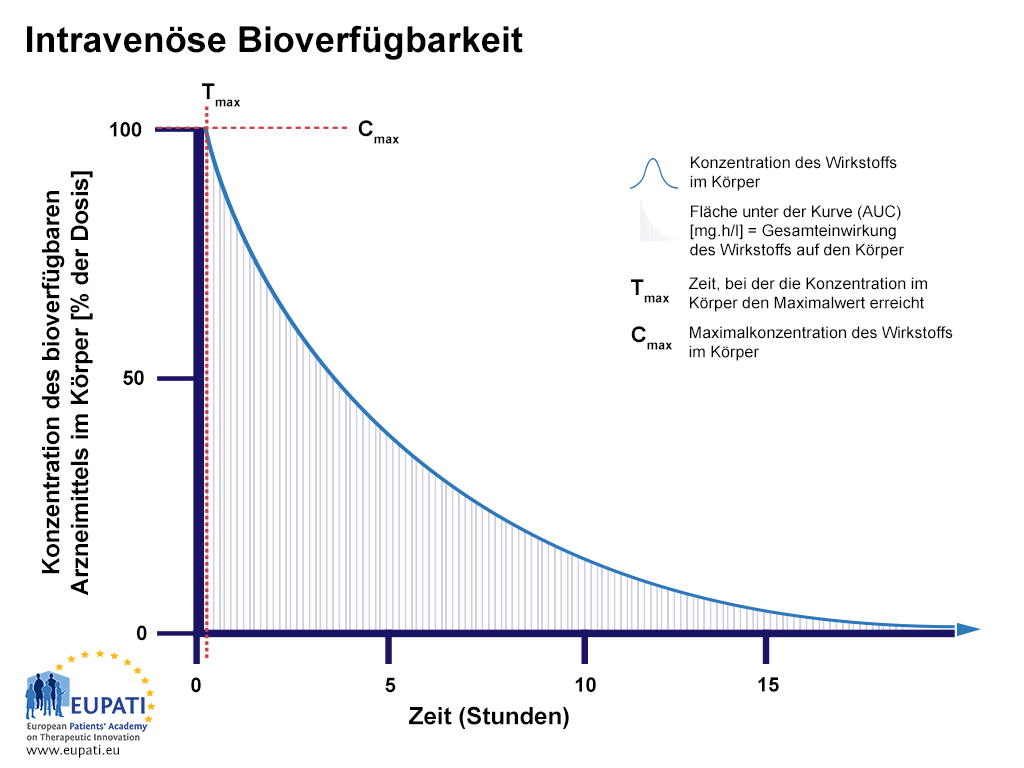
- Der Prozentsatz des Wirkstoffs im Körper oder die Bioverfügbarkeit direkt nach Injektion in die Blutbahn, untersucht über einen Zeitraum von 15 Stunden. Der Bereich unter der Kurve (engl. Area under the Curve; kurz AUC) ist schattiert dargestellt. Tmax bezeichnet den Zeitpunkt, zu dem die höchste Konzentration des Arzneimittels im Blut gemessen wird; Cmax bezeichnet die maximale Konzentration des Arzneimittels im Blut.
Bei einer Injektion erreicht der Wirkstoff nach einer komplexen Reise in der Blutbahn den Wirkort. Bei der Beurteilung der Bioverfügbarkeit werden Blutproben abgenommen und die Konzentration des Wirkstoffs im Blut (systemische Zirkulation) bestimmt. Unmittelbar nach der Injektion beträgt die Bioverfügbarkeit somit 100 %, da der Wirkstoff direkt in das Blut abgegeben wird. Genau dies können Sie der y-Achse der vorstehenden Abbildung (intravenöse Bioverfügbarkeit) entnehmen. Werden also 75 mg (Milligramm) des Wirkstoffs in die Blutbahn injiziert, entsprechen 100 % exakt 75 mg Wirkstoff.
Während der Wirkstoff in der Blutbahn zirkuliert, wird ein gewisser Anteil des Wirkstoffs verstoffwechselt oder ausgeschieden, wodurch die Konzentration des Wirkstoffs im Körper im Laufe der Zeit abnimmt (siehe vorstehende Abbildung). Um das Bioverfügbarkeitsprofil zu evaluieren und es mit dem anderer Arzneimittel zu vergleichen, wird die so genannte „Fläche unter der Kurve“ (AUC, Area under the curve) herangezogen; diese repräsentiert die Gesamtexposition des Körpers gegenüber einem Wirkstoff. Der Zeitpunkt, zu dem die höchste Konzentration des Wirkstoffs im Blut vorliegt, wird als Tmax bezeichnet, die maximale Konzentration des Wirkstoffs in der Blutbahn als Cmax.
Wird derselbe Wirkstoff wie in der vorstehenden Abbildung auf andere Weise verabreicht (beispielsweise durch orale Einnahme einer Tablette), beträgt die Bioverfügbarkeit weniger als 100 % (siehe nachstehende Abbildung „Orale Bioverfügbarkeit“).
-
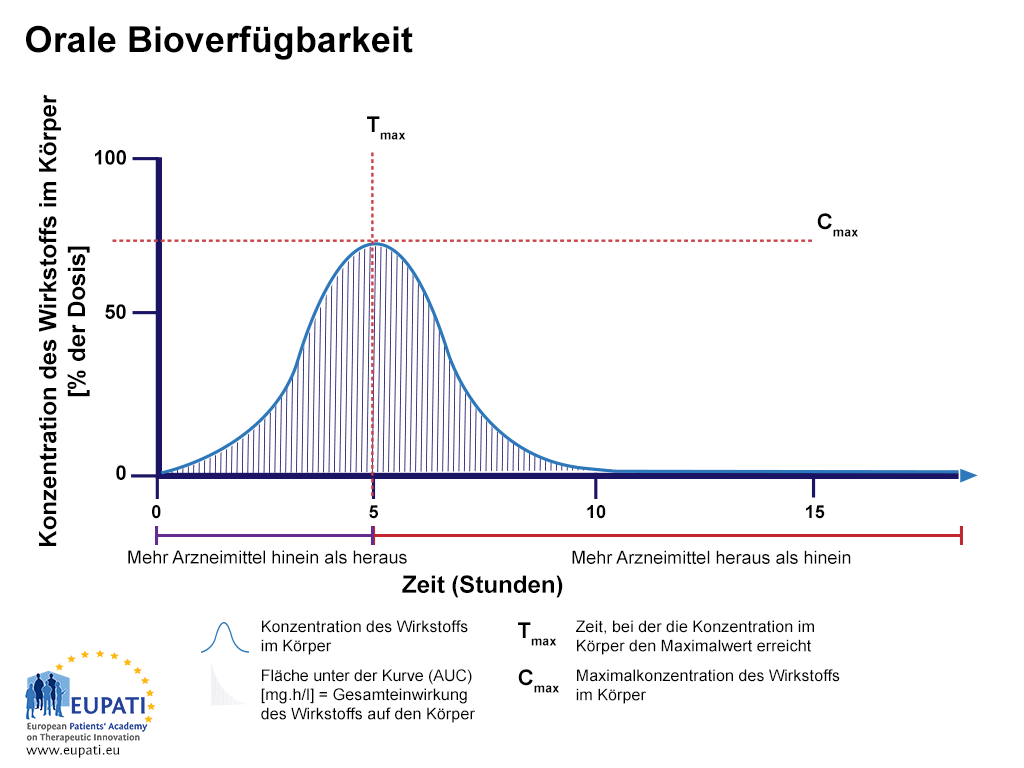
- Der Prozentsatz des Wirkstoffs nach Schlucken einer Tablette, untersucht über einen Zeitraum von 15 Stunden. Die Fläche unter der Kurve (AUC) ist schattiert dargestellt. Tmax bezeichnet den Zeitpunkt, zu dem die höchste Konzentration des Wirkstoffs im Blut gemessen wird, wohingegen Cmax die maximale Konzentration des Wirkstoffs in der Blutbahn bezeichnet.
Orale Bioverfügbarkeit
Der Prozentsatz des Wirkstoffs nach Schlucken einer Tablette, untersucht über einen Zeitraum von 15 Stunden. Die Fläche unter der Kurve (AUC) ist schattiert dargestellt. Tmax bezeichnet den Zeitpunkt, zu dem die höchste Konzentration des Wirkstoffs im Blut gemessen wird, wohingegen Cmax die maximale Konzentration des Wirkstoffs in der Blutbahn bezeichnet.
Die nachstehende Abbildung (Orale und intravenöse Bioverfügbarkeit) erläutert die geringere Bioverfügbarkeit bei oraler Verabreichung im Vergleich zur Verabreichung mittels intravenöser Injektion:
-
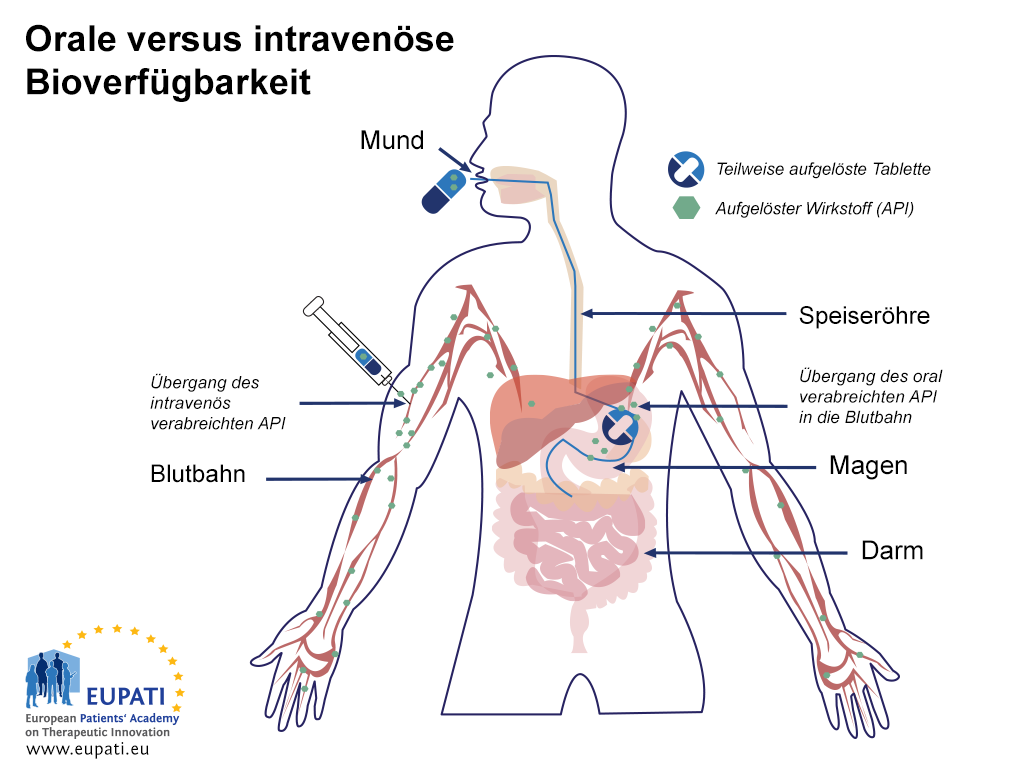
- Eine schematische Darstellung, welche die Resorption einer oral verabreichten Kapsel im Vergleich zu einer direkt in die Blutbahn verabreichten Injektion (intravenöse Injektion) zeigt. Nach Erreichen des Magens wird die Kapsel in den Dünndarm weitertransportiert, wo die weitere Resorption stattfindet.
Nachdem eine Tablette oder Kapsel geschluckt wurde, erreicht sie innerhalb von 1 bis 2 Minuten den Magen.1 Im Magen wird die Tablette oder Kapsel aufgelöst, und ein gewisser Teil des Wirkstoffs wird in die Blutbahn resorbiert. Die Komponenten gelangen in den Dünndarm, wo die Resorption abgeschlossen wird. Die Resorption im Gastrointestinalsystem kann stark variieren. Eine geringere Bioverfügbarkeit kann das Ergebnis einer schlechten oder gar nicht stattfindenden Resorption im Magen und im Darm sein, so dass es sich hierbei um einen wichtigen Schritt handelt, der die Verfügbarkeit beeinflussen kann.
Wenn der Wirkstoff resorbiert wird, gelangt er zuerst in die Leberpfortader und durch diese in die Leber. Dies ist das erste Mal, dass der Wirkstoff in der Leber verstoffwechselt wird; dies wird als „First-Pass-Verstoffwechselung“ bezeichnet. Manche Wirkstoffe werden bei dieser erstmaligen Verstoffwechselung in einem höheren Ausmaß verstoffwechselt als andere. Der nicht verstoffwechselte Anteil des Wirkstoffs – normalerweise weniger als 100 % – gelangt durch die Lebervene in die systemische Zirkulation. Die Menge, die tatsächlich in die systemische Zirkulation gelangt, wird als „absolute Bioverfügbarkeit“ bezeichnet.
Die absolute Bioverfügbarkeit vergleicht die Bioverfügbarkeit des Wirkstoffs in der systemischen Zirkulation nach nicht-intravenöser Verabreichung mit der Bioverfügbarkeit desselben Arzneimittels nach intravenöser Verabreichung. Es handelt sich somit also um den prozentualen Anteil des Wirkstoffs, der bei nicht-intravenöser Verabreichung resorbiert wird, bezogen auf den Wirkstoffspiegel bei intravenöser Verabreichung.
Kurz gesprochen stellt die intravenöse Verabreichung den Standard für die absolute Bioverfügbarkeit dar.
Die relative Bioverfügbarkeit bestimmt die Bioverfügbarkeit einer Formulierung (A) eines bestimmten Arzneimittels im Vergleich zu einer anderen Formulierung (B) des gleichen Arzneimittels, üblicherweise einem etablierten Standard (nicht: intravenöse Verabreichung) oder einer auf anderem Wege verabreichten Formulierung.
Die Bioverfügbarkeit wird von einer Vielzahl anderer, sich von Individuum zu Individuum unterscheidender Faktoren beeinflusst. Das beigefügte Datenblatt enthält einige Beispiele zur Bioverfügbarkeit.
Bioäquivalenz
Bioäquivalenz ist die Beziehung zwischen zwei Zubereitungen desselben Arzneimittels in der gleichen Darreichungsform, die eine vergleichbare Bioverfügbarkeit aufweisen.
Die relative Bioverfügbarkeit wird nicht nur für den Vergleich unterschiedlicher Formulierungen eingesetzt, sondern auch dann, wenn zwei Tabletten (oder beliebige andere Arzneimittel mit derselben Formulierung) mit demselben Wirkstoff von verschiedenen pharmazeutischen Unternehmen miteinander verglichen werden müssen. Die Tablette von Unternehmen A wird als ein Generikum der Referenz-Tablette von Unternehmen B (Markenarzneimittel) bezeichnet. Um festzustellen, ob eine Bioäquivalenz zwischen Tablette A und Tablette B besteht, werden die Bioverfügbarkeitsraten der beiden Tabletten miteinander verglichen.2
Weitergehende Informationen
- Food and Drug Administation (2002). Guidance for industry: Bioavailability and bioequivalence studies for orally administered drug products – General considerations. Rockville, MD: Food and Drug Administration. http://www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/AbbreviatedNewDrugApplicationANDAGenerics/UCM154838.pdf (Stand: 23. Juni 2015).
- Wang H, Li Q, Reyes S, Zhang J, Xie L, Melendez V, Hickman M, Kozar MP. (2013). Formulation and particle size reduction improve bioavailability of poorly water-soluble compounds with antimalarial activity. Malaria Research and Treatment, http://dx.doi.org/10.1155/2013/769234 (Stand: 23. Juni 2015).
- Johnson JA. (2000). Predictability of the effects of race or ethnicity on pharmacokinetics of drugs. International Journal of Clinical Pharmacology and Therapeutics, 38, 53-60.
Quellenangaben
- Tatum, R.P., Shi, G., Manka, M.A., Brasseur, J.G., Joehl, R.J. and Kahrilas, P.J. (2000). Bolus transit assessed by an esophageal stress test in postfundoplication dysphagia. Journal of Surgical Research, 91, 56–60.
- MobiSystems, Inc. (2007). Dorland's Medical Dictionary for Health Consumers. [Mobile application software].
Anlagen
- Datenblatt: Bioverfügbarkeit – Beispiele
Size: 100,543 bytes, Format: .docx
Beispiele für Bioverfügbarkeit bezogen auf Penicillin und Asthma.
A2-1.16-V1.2