Bases de la réglementation des médicaments
Pourquoi les médicaments sont-ils réglementés ?
Tout le monde souhaite pouvoir bénéficier d’un traitement médical en cas de maladie. Ainsi, des médicaments efficaces contre la maladie sont nécessaires.
Malheureusement, tous les médicaments s’accompagnent d’effets secondaires indésirables. Quoiqu’il en soit, les médicaments commercialisés doivent être sûrs pour un usage normal.
Les médicaments doivent être fiables. Cela signifie qu’ils doivent être fabriqués en se conformant à des normes de haute qualité.
Tous ces points sont traités via la réglementation. Les médicaments sont réglementés pour garantir que seuls des médicaments suffisamment sûrs, efficaces et de haute qualité puissent être commercialisés.
Qui réglemente les médicaments à l’échelle mondiale ?
Il n’existe pas de réglementation mondiale des médicaments. Cependant, depuis plus de 20 ans, des efforts ont été entrepris afin d’harmoniser la réglementation des médicaments à l’échelle internationale. Le Conseil international d’harmonisation (CIH) implique une collaboration entre les autorités réglementaires et l’industrie pharmaceutique de l’UE, des États-Unis, du Japon, du Canada, de Suisse et d’autres organisations régionales, avec l’Organisation mondiale de la santé (OMS) et un certain nombre d’autorités législatives ou administratives nationales qui tiennent un rôle d’observateurs.
Qui réglementent les médicaments dans l’Union européenne (UE) ?
Dans l’UE, la réglementation des médicaments est harmonisée entre tous les États membres selon un ensemble commun de règles législatives et elle implique la Commission européenne (CE), l’Agence européenne des médicaments (EMA) et les Autorités nationales compétentes (ANC) (autorités réglementaires). Ces règles couvrent également l’Espace économique européen (EEE), qui inclut la Norvège, l’Islande et le Liechtenstein. Ailleurs dans le monde, les médicaments sont réglementés au niveau national par l’autorité nationale compétente (ANC) de chaque pays, et l’harmonisation se fait entre les régions du CIH.
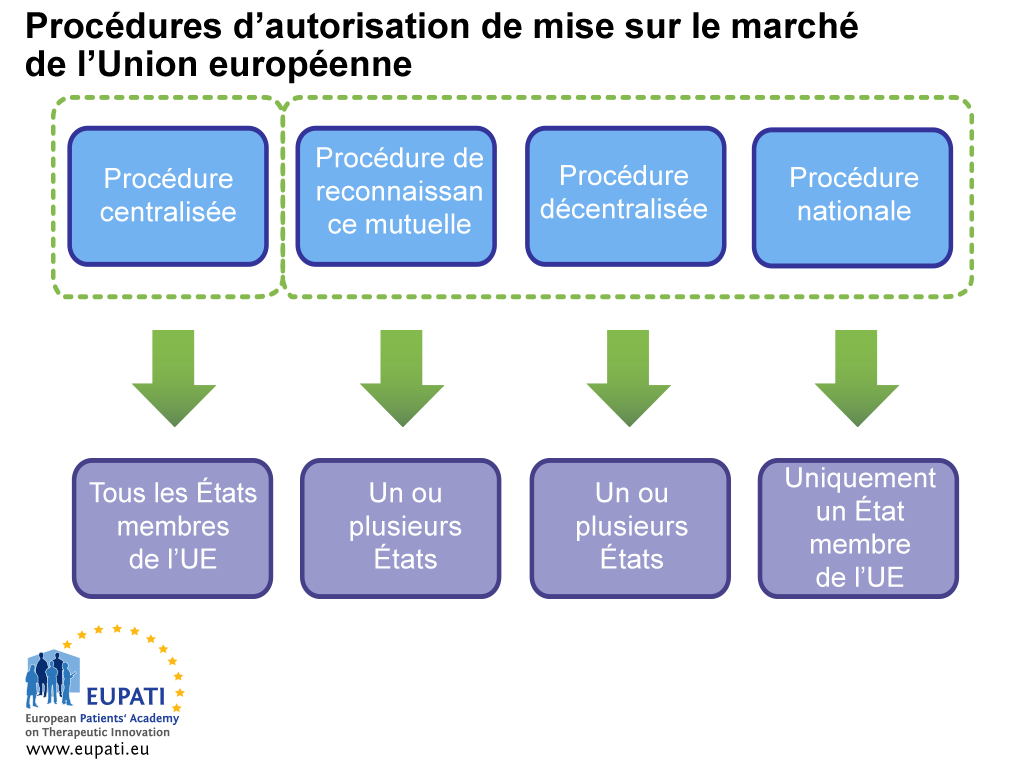
Du fait de cette harmonisation, les médicaments de l’UE peuvent être autorisés simultanément dans tous les pays de l’UE et de l’EEE via la procédure centralisée, qui est supervisée par l’EMA. Un médicament peut également être autorisé dans les États membres de l’UE via la procédure décentralisée (Decentralised Procedure – DCP), la procédure de reconnaissance mutuelle (Mutual Recognition Procedure – MRP) et la procédure nationale. Elles impliquent les autorités nationales compétentes (ANC) et ne s’appliquent pas automatiquement à tous les États membres européens.
-
- Différents acteurs sont impliqués dans l’autorisation de mise sur le marché d’un médicament selon la procédure choisie par le promoteur ou celle qu’il est obligé de suivre.
Le rôle de l'Agence européenne des médicaments dans la réglementation et l'agrément des médicaments
L'Agence européenne des médicaments (EMA) joue un rôle important dans la réglementation et l'agrément des médicaments.
L'Agence européenne des médicaments s'appuie sur les résultats d'essais cliniques réalisés par des établissements pharmaceutiques pour se former un avis quant à l'autorisation de médicaments. Elle gère également une base de données d'essais cliniques réalisés dans l'Union européenne https://www.clinicaltrialsregister.eu/
L'EMA supervise la procédure centralisée (PC) pour l'autorisation de mise sur le marché (AMM). Dans l'ensemble, les nouveaux médicaments sollicitent une autorisation via la PC. Pour cela, une société doit soumettre une demande unique à l'EMA et, si le médicament est agréé, alors une licence est accordée par la CE pour sa mise sur le marché dans tous les pays de l'UE et de l'EEE.
Le comité de l'EMA responsable de l'évaluation des dossiers (demandes) est le Comité des médicaments à usage humain (CHMP). Les experts qui évaluent les demandes sont nommés par chacun des États membres plus l'Islande et la Norvège. Ils peuvent être appuyés par un maximum de cinq membres cooptés, choisis parmi des experts nommés par les États membres ou l'EMA et sont recrutés, lorsque c'est nécessaire, pour fournir une expertise complémentaire dans un domaine scientifique spécifique.
A2-5.01.1-V1.1