Autorizzazione all’immissione in commercio
Introduzione
Tutti i farmaci devono avere un’autorizzazione all’immissione in commercio (Marketing Authorisation, MA) per essere immessi legalmente sul mercato nello Spazio economico europeo (SEE).1 L’obiettivo ultimo dell’autorizzazione all’immissione al commercio è di garantire la possibilità di rendere velocemente disponibili ai cittadini farmaci sicuri, efficaci e di elevata qualità. È importante accennare che i dispositivi medici devono seguire processi differenti rispetto agli altri prodotti medicinali, prima che siano messi a disposizione sul mercato.
Le autorità nazionali competenti (National Competent Authorities, NCA), in altre parole le autorità di regolamentazione, sono agenzie governative nazionali responsabili di valutare le richieste di autorizzazione all’immissione in commercio (Marketing Authorisation Applications, MAA) e di garantire le MA per i prodotti medicinali che sono immessi nei rispettivi mercati attraverso procedure nazionali, decentralizzate o di mutuo riconoscimento. L’Agenzia europea per i medicinali (European Medicines Agency, EMA) di Londra è responsabile per il coordinamento della valutazione scientifica delle richieste per MA a livello europeo per prodotti medicinali secondo la procedura centralizzata (Centralised Procedure, CP). La valutazione viene svolta da esperti delle NCA. Dopo una valutazione positiva, la Commissione europea emette la MA.
Procedure di autorizzazione all’immissione in commercio
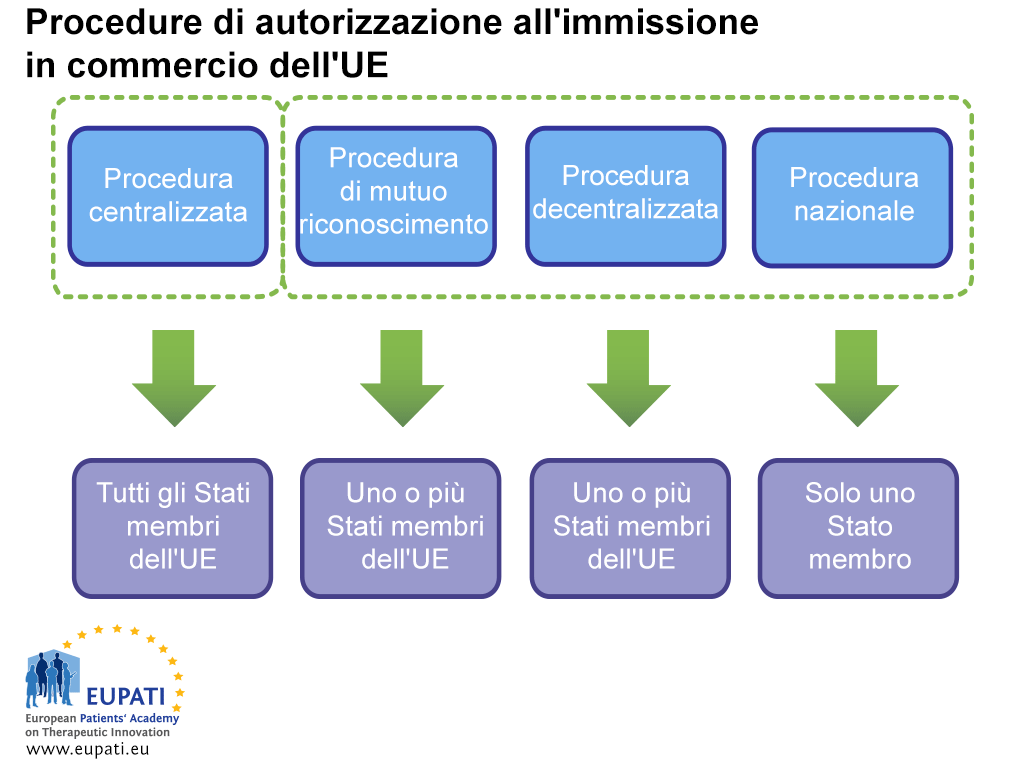
Vi sono due percorsi per il conseguimento di una MA: tramite la procedura centralizzata (CP) o tramite la procedura non centralizzata. Quest’ultima prevede la procedura decentralizzata (Decentralised Procedure, DCP), la procedura di mutuo riconoscimento (Mutual Recognition Procedure, MRP) e la procedura nazionale (National Procedure). Ciascun sistema ha le proprie disposizioni normative e le proprie responsabilità rispetto all’autorità competente (EMA o NCA) e ai titolari della MA.
Procedura centralizzata (CP)
La maggior parte dei nuovi farmaci prende questo percorso. L’azienda presenta una singola domanda all’EMA. Il Comitato per i prodotti medicinali ad uso umano (Committee for Human Medicinal Products, CHMP) dell’EMA, che è composto da un membro nominato dagli Stati membri dell’UE e dai paesi del SEE e cinque scienziati esperti, consigliano alla Commissione europea se un farmaco deve essere approvato, Se un farmaco viene approvato in questo modo, viene concesso un brevetto per la commercializzazione dei medicinali per tutti i paesi dell’UE e del SEE. Questo processo di valutazione scientifica richiede un massimo di 210 giorni attivi, ma questo conteggio può essere interrotto (chiamato “clock stop”).
Procedura decentralizzata (DCP) e procedura di mutuo riconoscimento (MRP).
Con tali procedure, vengono richieste autorizzazioni all’immissione in commercio in diversi Stati membri allo stesso tempo, con un paese avente il ruolo di guida in qualità di Stato membro di riferimento (Reference Member State, RMS). Se ha successo, il medicinale viene approvato per la commercializzazione nei RMS e altri paesi interessati (Concerned Member States, CMS). Tali procedure si basano sul principio del riconoscimento della valutazione dei RMS da parte dei CMS. La maggior parte dei farmaci generici sono approvati tramite DCP.
Procedura nazionale
Le procedure indipendenti nazionali sono limitate rigidamente ai farmaci che devono essere autorizzate e commercializzate solo in uno Stato membro (Member State, MS). Attualmente, per i nuovi prodotti tale procedura viene seguita di rado.
Selezione di una procedura di autorizzazione all’immissione in commercio
Un’azienda farmaceutica è di solito libera di decidere quale procedura utilizzare. Si tratta di una decisione economica che può includere vari motivi per la scelta di una procedura specifica. La CP è obbligatoria per:
- prodotti derivati da processi biotecnologici;
- farmaci per terapia avanzata (come la terapia genica, la terapia con cellule somatiche o farmaci basati sull’ingegneria tissutale);
- prodotti medicinali orfani;
- medicinali per il trattamento del virus dell’immunodeficienza umana (HIV) o della sindrome da immunodeficienza acquisita (AIDS), del cancro, di patologie neurodegenerative, disfunzioni autoimmuni o di altro tipo, nonché di malattie virali o diabete.
La CP può essere utilizzata anche per farmaci che rappresentano un’innovazione terapeutica, scientifica o tecnica rilevante o d’interesse per la salute pubblica, come medicinali per il calo ponderale.
Se un’azienda desidera sviluppare un farmaco esistente con un’autorizzazione all’immissione in commercio per un’indicazione diversa, conseguentemente devono di solito fare domanda per un’autorizzazione completamente nuova.
-
- In funzione della procedura che lo sponsor sceglie o è obbligato a seguire, vi sono diverse parti coinvolte nell’autorizzazione all’immissione in commercio di un farmaco.
Valutazione scientifica di farmaci
In merito alla procedura, ciascun farmaco necessita di un "dossier" standard per la valutazione dell'autorizzazione all'immissione in commercio. Il documento tecnico comune (Common Technical Document, CTD) è un formato riconosciuto a livello internazionale che deve essere seguito per le richieste da presentare alle autorità di regolamentazione. IL CTD è costituito da un insieme di documenti in cui il richiedente deve dimostrare che il prodotto medicinale ha la qualità necessarie e che è sicuro ed efficace.
Rifiuto di un'autorizzazione all'immissione in commercio
Una MA sarà rifiutata, nel caso di un prodotto medicinale:
- il cui bilancio rischi-benefici non è considerato favorevole;
- la cui efficacia terapeutica non è comprovata in modo sufficiente dal richiedente;
- la cui composizione qualitativa e quantitativa non sono quelle dichiarate.
Validità dell'autorizzazione all'immissione in commercio e follow up
La MA viene rinnovata dopo cinque anni sulla base di un'ulteriore valutazione del bilancio rischi-benefici dell'autorità nazionale competente di uno Stato membro autorizzante. Una volta rinnovata, la MA è valida per un periodo illimitato a meno che l'autorità competente non decida su basi giustificate relative alla farmacovigilanza di procedere a un rinnovo supplementare di cinque anni.
A tutti i titolari dell'autorizzazione all'immissione in commercio (marketing authorisation holder, MAH) è richiesto di fornire i dati sulla sicurezza al follow up e di seguire altri requisiti relativi al periodo di post-commercializzazione. Una volta immessi sul mercato, i farmaci devono in effetti continuare a essere monitorati (farmacovigilanza) al fine di garantire che qualsiasi aspetto che potrebbe incidere sul profilo di sicurezza di un medicinale sia individuato ed esaminato e che vengano prese le misure necessarie.
Mentre sono sul mercato, possono diventare disponibili nuovi dati o informazioni sull'efficacia, la sicurezza o la produzione del farmaco. In questo caso, è responsabilità del MAH inviare una richiesta di "variazione" di un'autorizzazione all'immissione in commercio valida alla/alle autorità competente/i in tutti gli MS che hanno autorizzato in precedenza il prodotto medicinale.
Il coinvolgimento dei pazienti nell'autorizzazione all'immissione in commercio
Il coinvolgimento dei pazienti nell'autorizzazione all'immissione in commercio è stata tradizionalmente molto limitata. Alcune autorità competenti, comunque, stanno pensando a come coinvolgere i pazienti nelle procedure di autorizzazione all'immissione in commercio: ad esempio, tramite la consultazione dei pazienti e/o una rappresentanza nella Commissione sui farmaci umani nell'ambito dell'Agenzia di regolamentazione dei farmaci e dei prodotti sanitari (Medicines and Healthcare products Regulatory Agency, MHRA) nel Regno Unito. I pazienti sono già coinvolti nelle discussioni sui rischi-benefici presso l'EMA come membri del Comitato di farmacovigilanza (Pharmacovigilance Committee, PRAC), attraverso la partecipazione alle procedure di supporto relative a raccomandazioni/protocolli scientifici. Esiste inoltre un'iniziativa pilota che include i pazienti nel CHMP durante le dichiarazioni orali rilasciate prima del processo decisionale.
I difensori dei diritti dei pazienti hanno inoltre la possibilità di influenzare la normativa nazionale/dell'UE riguardante l'autorizzazione all'immissione in commercio.
Riferimenti bibliografici
- The European Economic Area (EEA) unites the EU Member States and the three EEA EFTA States (Iceland, Liechtenstein, and Norway) into the internal market governed by the same basic rules. These rules aim to enable goods, services, capital, and persons to move freely about the EEA in an open and competitive environment, a concept referred to as the four freedoms.
Allegati
A2-5.07-v1.1