Anträge auf Marktzulassung
Einleitung
Die Arzneimittelentwicklung ist ein langer Prozess. Das ultimative Ziel des Arzneimittelentwicklungsprozesses ist eine Genehmigung für die Vermarktung des neuen Arzneimittels – eine Marktzulassung. Innerhalb von Pharmaunternehmen sind (oder sollten) Abteilungen für Zulassungsrechtliche Angelegenheiten ein integraler Bestandteil aller Schritte während des gesamten Lebenszyklus des Arzneimittels (sein). Diese Abteilungen sind insbesondere für die Anträge, die vor jeder klinischen Studie eingereicht werden müssen, die Vorbereitung und Einreichung des Dossiers für die Marktzulassung sowie andere Aktivitäten, nachdem die Marktzulassung erteilt wurde, zum Beispiel den Antrag auf eine Änderung der Marktzulassung (eine Variation) zuständig. Diese Fachleute müssen gründliche Kenntnisse sämtlicher anwendbaren Arzneimittelregelungen sowie des gesamten Entwicklungsprozesses besitzen.
-
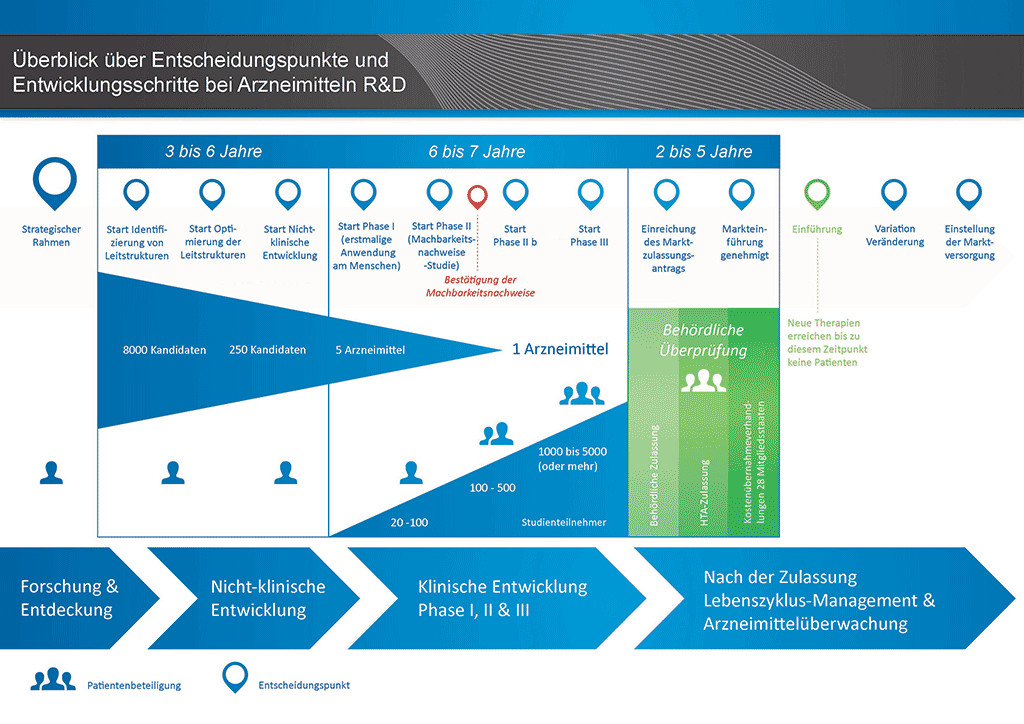
- Es benötigt mehr als 10 Jahre sorgfältiger Planung und Forschung, bis ein Arzneimittel sich vom Molekül zur marktfähigen Behandlung entwickelt hat.
Abbildung 1 – Überblick über den Arzneimittelentwicklungsprozess
Einreichungen für Marktzulassung (MA)
Das Pharmaunternehmen muss in einem frühen Stadium der Entwicklung entscheiden, welche Art von Antrag es für die Marktzulassung vorlegen möchte, z. B.:
- Einen vollständigen Antrag – siehe Common Technical Document (CTD) Dreieck unten.
- Einen verkürzten Antrag (reduzierter Antrag).
- Einen bibliografischen Antrag – basierend auf vorhandener wissenschaftlicher Literatur.
Für Anträge muss den zuständigen Behörden ein Dokumentationsdossier vorgelegt werden. Abbildung 1 zeigt den Entwicklungsprozess eines neuen, innovativen Arzneimittels. Für diese Art von Arzneimittel müsste ein vollständiges Dossier vorgelegt werden, in dem alle Elemente der Dokumentation für das Arzneimittel enthalten sein müssen.
Welche Elemente enthält ein Dossier?
Abbildung 2 zeigt die Elemente, aus denen das Common Technical Document (CTD) – das Dossier, welches den Zulassungsbehörden in Kanada, Europa, Japan, der Schweiz, den Vereinigten Staaten und anderen als Antrag auf Marktzulassung (MAA) vorgelegt wird – zusammengestellt ist. Das Format des CTD wurde vom Internationalen Rat zur Harmonisierung der Beurteilungskriterien von Human-Arzneimitteln (ICH) entwickelt. Das CTD muss unabhängig von dem Verfahren (zentralisiertes Verfahren [CP], Mutual Recognition Procedure [MRP], dezentralisiertes Verfahren [DCP] oder nationales Verfahren [NP]) oder der Art des Antrags für alle Arten von Marktzulassungsanträgen in der EU verwendet werden (Stand-alone, Generika, usw.). Das CTD-Format gilt für alle Produkttypen (neue Wirksubstanzen, Radiopharmaka, Impfstoffe, pflanzliche Arzneimittel, usw.).
Im CTD gibt es fünf unterschiedliche Module.
- Modul 1: Regionale administrative Informationen.
- Modul 2: Zusammenfassungen und Übersichten.
- Modul 3: Qualität.
- Modul 4: Berichte aus vorklinischen Prüfungen.
- Modul 5: Berichte aus klinischen Prüfungen.
Module 2 bis 5 des CTD sind für alle Regionen gleich, während Modul 1 für jede Region spezifisch ist und wird nicht als Bestandteil des CTD betrachtet. Die Mehrheit der Dokumentation zu Qualität, Sicherheit und Wirksamkeit des Arzneimittels sind in den Modulen 3 bis 5 enthalten. Zulassungsrechtliche Fachleute stellen sicher, dass die vorgelegte Dokumentation mit sämtlichen relevanten Verordnungen, Richtlinien und Leitlinien im Einklang ist. Sie erstellen auch die Zusammenfassungen, die im Modul 2 enthalten sind.
Abbildung 2: Das Common Technical Document (CTD) Dreieck.
Modul 1: Regionale administrative Informationen
Modul 1 des CTD enthält sämtliche, auf regionaler Ebene erforderlichen administrativen Informationen. Die EU besitzt ihre eigene Version von Modul 1. Es besteht aus den folgenden 10 Elementen:
1.0 Anschreiben
1.1 Umfassendes Inhaltsverzeichnis
1.2 Antragsformular
1.3 Produktinformationen
Das sind Informationen, die sowohl von Angehörigen der Gesundheitsberufe als auch von Patienten verwendet werden. Enthalten sind die Zusammenfassung der Merkmale des Arzneimittels – ein detailliertes Dokument, das sich an Angehörige der Gesundheitsberufe richtet, Etikettierung sowie die Packungsbeilage. Es muss ein Lesbarkeitstest durchgeführt werden, um zu zeigen, dass die Packungsbeilage für Laien verständlich ist. Die zuständigen nationalen Behörden und die EMA haben Vorlagen in allen EU-Sprachen für die Präsentation von Produktinformationen veröffentlicht, die das Format und den Inhalt im Detail vorschreiben.
1.4 Informationen über die Experten
Modul 2 des CTD enthält von Experten verfasste Zusammenfassungen und Übersichten. Jeder dieser Experten sollte einen Lebenslauf (CV) zur Verfügung stellen und eine Erklärung unterzeichnen, dass sie bei der Erstellung der Zusammenfassungen die Regeln der geltenden Verordnungen oder Richtlinien befolgt haben.
1.5 Spezifische Anforderungen für verschiedene Arten von Anträgen
Für spezielle Arten von Anträgen sind zusätzliche Informationen erforderlich, wie bibliografische Anträge, generische, „hybrid” oder bio-ähnliche Anträge, (erweiterte) Daten/Marktexklusivität, Antrag unter außergewöhnlichen Umständen oder Antrag auf eine bedingte Marktzulassung.
1.6 Umgebungsbedingte Risikobewertung
Alle Wirkstoffe in Arzneimitteln können ein potenzielles Risiko für die Umwelt darstellen, und alle Stoffe oder deren Metaboliten gelangen letztendlich in die Umwelt. Das Unternehmen muss auf die möglichen Auswirkungen auf die Umwelt durch Verwendung, Lagerung und Entsorgung des Arzneimittels eingehen.
1.7 Informationen zu Marktexklusivität für Arzneimittel für seltene Leiden
Spezielle Informationen sind erforderlich, wenn mit dem als Arzneimittel für seltene Leiden bezeichneten Arzneimittel eine seltene Erkrankung behandelt werden soll. Sofern ein anderes, auf dem Markt verfügbares Arzneimittel über Marktexklusivität für die gleiche Indikation verfügt, kann das neue Arzneimittel nur unter besonderen Bedingungen zugelassen werden.
1.8 Informationen zu Pharmakovigilanz
Es muss eine Beschreibung der Pharmakovigilanz und Risikomanagementsysteme enthalten sein. Der Antragsteller muss nachweisen, dass eine ordnungsgemäße Überwachung von Nebenwirkungen und möglichen Risiken vorhanden ist. Dies sollte einen Nachweis enthalten, dass der Antragsteller über eine qualifizierte Person, die für die Pharmakovigilanz verantwortlich ist, sowie über die notwendigen Mittel für die Anmeldung von auftretenden Nebenwirkungen entweder in der EU oder in einem Drittland (Artikel 8 (n) der Richtlinie 2001/83/EG) verfügt.
1.9 Informationen zu klinischen Studien
Der Antragsteller muss eine Erklärung beifügen, dass sämtliche, mit dem Arzneimittel außerhalb der EU durchgeführten klinischen Studien die EU-Anforderungen erfüllen.
1.10 Informationen zu Pädiatrie
In der EU müssen sämtliche neuen Arzneimittel für die pädiatrische Bevölkerungsgruppe berücksichtigt werden. Im Allgemeinen sollten sie bei Kindern getestet werden. Jedoch kann eine Befreiung von dieser Anforderung gewährt werden, wenn die Erkrankung nur bei älteren Menschen oder Erwachsenen vorkommt, oder wenn das neue Arzneimittel in der gesamten oder einem Teil der pädiatrischen Population möglicherweise unwirksam oder bedenklich ist. Wird keine Befreiung erteilt, so muss das Unternehmen ein Pädiatrisches Prüfkonzept (PIP - Paediatric Investigation Plan) vorlegen, es sei denn, es wurde eine Verschiebung gewährt, sodass das PIP später erstellt werden kann. Dieser Abschnitt sollte eine Kopie der Befreiung oder der Entscheidung zum PIP (einschließlich Verschiebungen, falls zutreffend) enthalten.
Wie wird das Dossier zusammengestellt?
In den meisten Fällen sind Einreichungen in Papierform nicht mehr möglich, was bedeutet, dass die gesamte Dokumentation innerhalb der fünf CTD-Module in einem standardisierten elektronischen Format, der eCTD vorliegen muss. Die eCTD ist weder eine Sammlung von PDF-Dokumenten, noch eine einzige große PDF-Datei. Vielmehr ist die eCTD ein Standard, der die Verzeichnis- und Dateistruktur, die befolgt werden muss, detailliert beschreibt, damit das Unternehmen und die Zulassungsbehörden im Dossier leicht navigieren können, wie in einem gewöhnlichen Computerverzeichnis.
Was ist der Einreichungsprozess?
Der Antragsteller muss sämtliche logistischen und zulassungsrechtlichen Fragen vor der Einreichung sorgfältig prüfen. Dazu zählt die Wahl des anzuwendenden Marktzulassungsverfahrens: das zentralisierte Verfahren (CP), das Verfahren der gegenseitigen Anerkennung (MRP), das dezentralisierte Verfahren (DCP) oder das nationale Verfahren (NP).
-
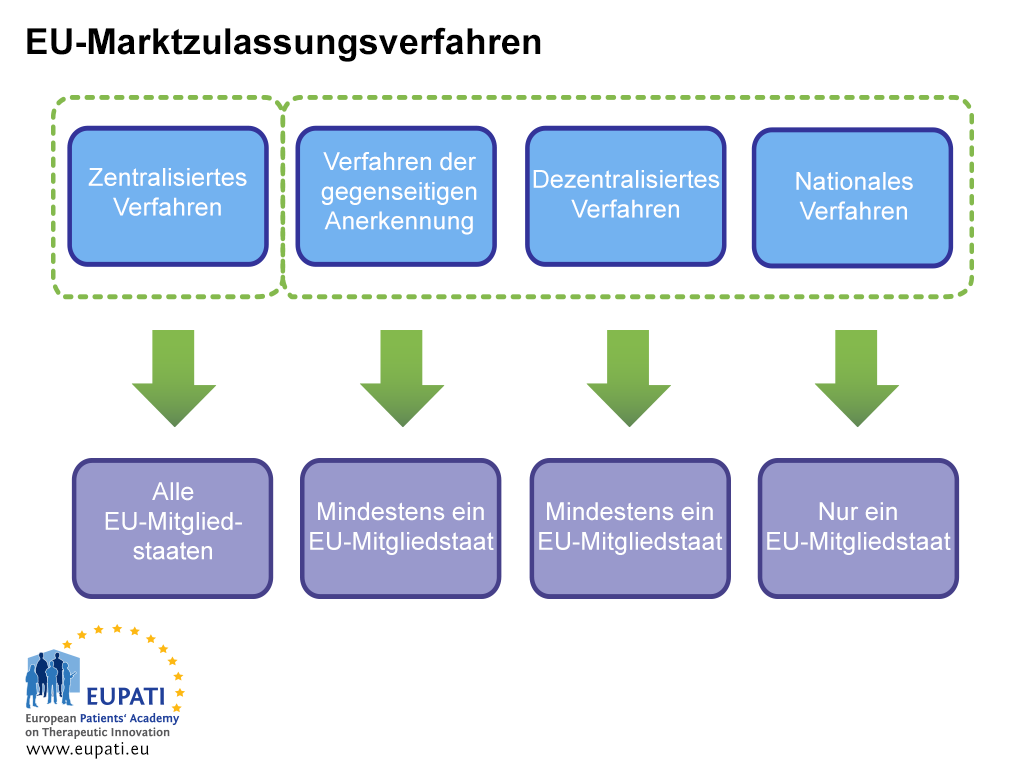
- An der Marktzulassung eines Arzneimittels sind verschiedene Parteien beteiligt, je nachdem, welches Verfahren der Sponsor für die Beantragung ausgewählt hat (oder befolgen muss).
Antworten auf viele Fragen betreffend Anträge im Wege des CP sind auf der EMA-Website zu finden.
Treffen vor der Einreichung
Treffen zwischen dem Unternehmen und Mitarbeitern der Zulassungsbehörde vor der Einreichung finden im Allgemeinen sechs bis sieben Monate vorm Datum der Einreichung statt. Diese Treffen werden organisiert, damit das Unternehmen weitere Informationen und Anleitungen erhalten kann, bevor das Antragsdossier fertiggestellt wird.
Im Falle von Marktzulassungsanträgen nach dem CP trifft das Projektteam des Unternehmens das Team der EMA, das in die Bewertung des Antrages involviert ist. Bei MRP, DCP oder NP sind Treffen mit den zuständigen nationalen Behörden vor der Einreichung möglich und gleichermaßen nützlich.
Einreichung des Antrags auf Marktzulassung (Marketing Authorisation Application, MAA)
Im CP sind Einreichungen nur an die EMA im eCTD-Format möglich, es sei denn, es wurde eine Ausnahme gewährt. Die eCTD wird über ein Online-Portal eingereicht.
Im Falle von Anträgen nach dem MRP, DCP oder NP ist die Situation etwas komplizierter. Bei diesen Anträgen können bis zu 31 verschiedene Agenturen involviert sein. Das HMA-Netzwerk (Heads of Medicines Agencies –Leiter der Arzneimittelagenturen, eine Zusammenarbeit zwischen allen Mitgliedstaaten) bietet nun eine der EMA ähnliche Lösung: die Gemeinsame Europäische Einreichungs-Plattform (CESP, Common European Submission Platform). Die CESP zu verwenden bedeutet, dass das Unternehmen ein Dossier lediglich einmal in das System hochladen muss; danach können alle beteiligten Mitgliedstaaten die Einreichung aus dem CESP-Speicher beziehen. Die Plattform ermöglicht auch die Kommunikation zwischen Behörden und dem Antragsteller.
Die Validierungsphase
Sobald die EMA oder die zuständige nationale Behörde den Antrag auf Marktzulassung erhält, wird das Dossier zunächst überprüft, um sicherzustellen, dass sämtliche erforderlichen Dokumente enthalten sind. Bei Fragen besteht für den Antragsteller die Möglichkeit, die erforderlichen Antworten und zusätzliche Unterlagen einzureichen. Sobald der Antrag auf Marktzulassung überprüft wurde, beginnt die Bewertung.
A2-5.11-V1.1