Ansøgninger om markedsføringstilladelse
Introduktion
Udvikling af lægemidler er en langvarig proces. Det endelige mål for enhver udviklingsproces er godkendelse af det nye lægemiddel til markedsføring – en markedsføringstilladelse (MA – Marketing Authorisation). I lægemiddelvirksomheder indgår afdelingen for regulatoriske forhold i alle trin i et lægemiddels livscyklus, og hvis det ikke er tilfældet, burde den gøre det. Afdelingen for regulatoriske forhold har især ansvaret for de ansøgninger, der skal indsendes før hvert klinisk forsøg, udarbejdelse og indsendelse af dossieret vedr. ansøgningen om markedsføringstilladelse og andre opgaver efter bevilling af markedsføringstilladelsen, f.eks. ansøgning om ændring af markedsføringstilladelsen. Specialister i regulatoriske forhold må have et omfattende kendskab til alle gældende regler om lægemidler og til hele udviklingsprocessen.
-
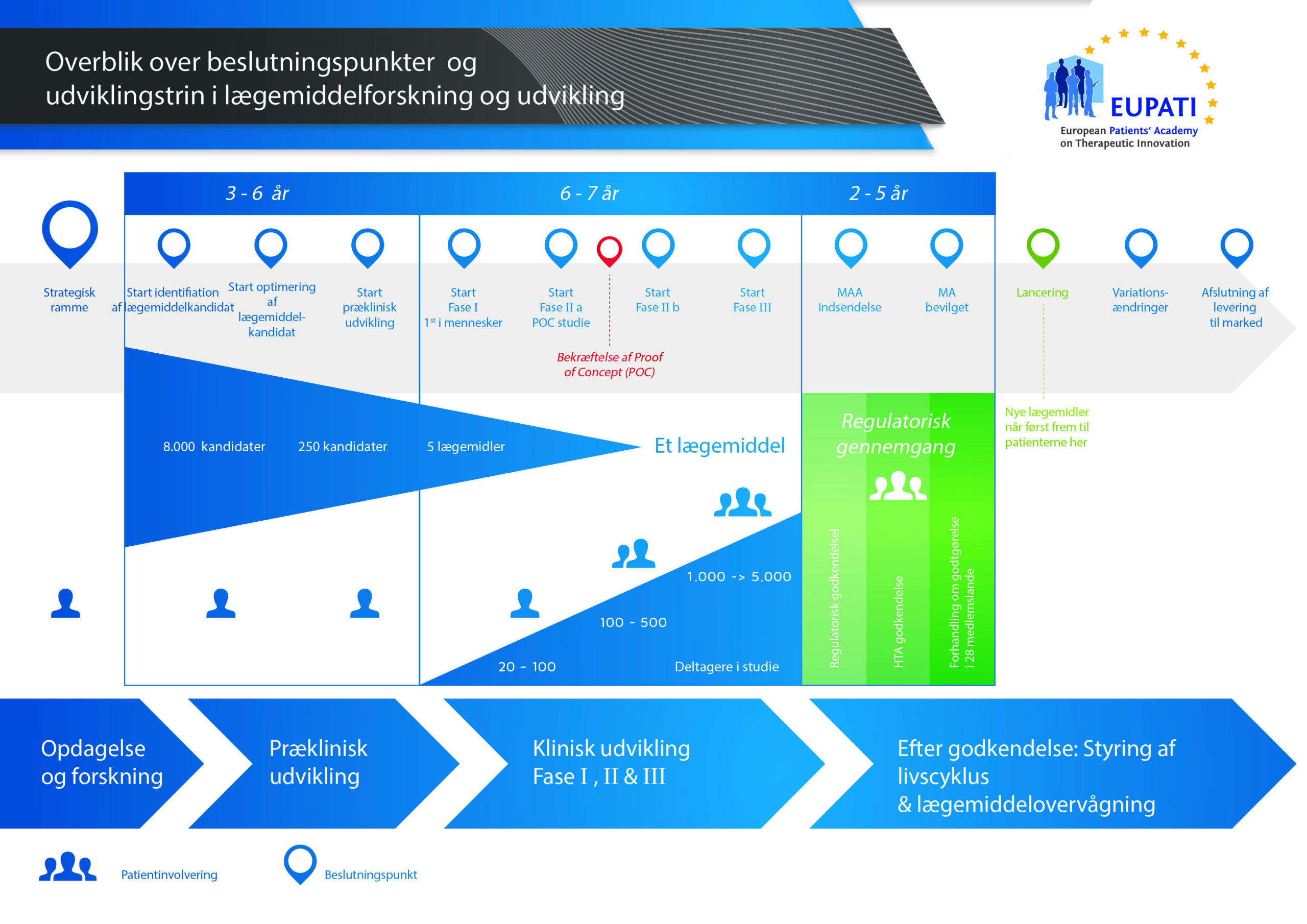
- Det kræver langt over 10 års omhyggelig planlægning og forskning, før et lægemiddel kan tages fra molekyle til behandling, der er klar til at komme på markedet.
Indsendelse af ansøgning om markedsføringstilladelse (MA – Marketing Authorisation)
Lægemiddelvirksomheden skal på et tidligt stadium beslutte, hvilken type ansøgning om markedsføringstilladelse der skal indsendes, f.eks.:
- En komplet ansøgning – se Fælles Teknisk Dokument (FTD) nedenfor.
- En forkortet ansøgning.
- En bibliografisk ansøgning – baseret på eksisterende videnskabelig litteratur.
I forbindelse med en ansøgning skal der indsendes et dokumentationsdossier til de relevante myndigheder. Figur 1 viser udviklingsprocessen for et nyt, innovativt lægemiddel. Denne type lægemiddel kræver, at der indsendes et komplet dossier, som skal omfatte alle elementer af dokumentationen for lægemidlet.
Hvilke elementer skal et dossier indeholde?
Figur 2 viser de elementer, der udgør det Fælles Tekniske Dokument (FTD), dvs. det dossier, der indsendes til lægemiddelmyndighederne ved ansøgning om markedsføringstilladelse (MAA – Marketing Authorisation Application) i Canada, Europa, Japan, Schweiz og USA med flere. Formatet for det Fælles Tekniske Dokument (FTD) er udviklet af det internationale råd for harmonisering af tekniske krav til medicinske lægemidler til mennesker (ICH – International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use). FTD bruges til alle typer MAA'er i EU, uanset procedure (central procedure CP, gensidig anerkendelsesprocedure MRP (Mutual Recognition Procedure), decentral procedure DCP eller national procedure NP) og ansøgningstype (separat, generisk osv.). FTD-formatet gælder for alle typer produkter (nye kemiske enheder, radiofarmaceutiske midler, vacciner, urtemedicin osv.).
FTD indeholder fem forskellige moduler.
- Modul 1: Regionale administrative oplysninger.
- Modul 2: Resuméer og oversigter.
- Modul 3: Kvalitet.
- Modul 4: Rapporter fra non-kliniske undersøgelser.
- Modul 5: rapporter for kliniske undersøgelser.
FTD-modul 2 til 5 er fælles for alle områder, mens modul 1 er specifikt for hvert enkelt område og anses ikke for at være en del af FTD. Hovedparten af den dokumentation, der vedrører lægemidlets kvalitet, sikkerhed og effekt, findes i modul 3 til 5. Specialister i regulatoriske forhold sørger for, at den indsendte dokumentation overholder alle relevante forordninger, direktiver og retningslinjer. De udarbejder også de resuméer, der indgår i modul 2.
-

- Præklinisk udvikling i CTD-moduler. Udarbejdet fra ICH’s fælles tekniske dokument (se reference 1)
Figur 2: Fælles teknisk dokument (FTD) – trekanten.
Modul 1: Regionale administrative oplysninger
FTD-modul 1 indeholder alle de administrative oplysninger, der er nødvendige på regionalt niveau. EU har sin egen version af modul 1. Den består af følgende 10 elementer:
1.0 Følgebrev
1.1 Udførlig indholdsfortegnelse
1.2 Ansøgningsformular
1.3 Produktoplysninger
Disse oplysninger vil blive anvendt af både sundhedspersonale og patienter. De omfatter produktresuméet – et udførligt dokument til sundhedspersonale, etikettering og indlægssedlen. En læsbarhedstest skal gennemføres, så det kan påvises, at indlægssedlen er forståelig for lægfolk. De nationale kompetente myndigheder og EMA har offentliggjort skabeloner på alle EU-sprog til præsentation af produktoplysninger, som i detaljer fastlægger formatet og indholdet.
1.4 Oplysninger om eksperterne
FTD-modul 2 indeholder resuméer og oversigter, der er skrevet af eksperter. Hver af disse eksperter skal levere sit CV og underskrive en erklæring om, at vedkommende har fulgt reglerne i gældende forordninger og direktiver under udarbejdelsen af resuméerne.
1.5 Specifikke krav til forskellige typer af ansøgninger
Der kræves yderligere oplysninger til særlige typer af ansøgninger, f.eks. bibliografiske ansøgninger, generiske, hybrid- eller biosimilære ansøgninger, (udvidet) data-/markedseneret, ansøgning under særlige omstændigheder eller ansøgning om betinget markedsføringstilladelse.
1.6 Vurdering af miljørisici
Alle aktive stoffer i lægemidler kan udgøre en potentiel risiko for miljøet, og alle stoffer eller deres metabolitter havner i sidste ende i miljøet. Virksomheden skal forholde sig til den mulige miljøpåvirkning, som brugen, opbevaringen og bortskaffelsen af lægemidlet indebærer.
1.7 Oplysninger vedrørende markedseneret for lægemidler til sjældne sygdomme
Der kræves særlige oplysninger, hvis lægemidlet er beregnet til behandling af en sjælden sygdom. Hvis et andet lægemiddel på markedet allerede har markedseneret for den samme indikation, kan det nye lægemiddel kun godkendes under særlige omstændigheder.
1.8 Oplysninger vedrørende lægemiddelovervågning
Der skal være en beskrivelse af systemerne til lægemiddelovervågning og risikostyring. Ansøgeren skal påvise, at der er etableret passende overvågning af bivirkninger og potentielle risici. Dette skal omfatte bevis for, at ansøgeren har en sagkyndig person, der er ansvarlig for lægemiddelovervågning, samt det nødvendige udstyr til at rapportere om enhver formodet bivirkning i EU eller et tredjeland (artikel 8, litra n) i direktiv 2001/83/EF).
1.9 Oplysninger vedrørende kliniske forsøg
Ansøgeren skal vedlægge en erklæring om, at kliniske forsøg gennemført uden for EU opfylder EU's krav.
1.10 Oplysninger vedrørende lægemidler til behandling af børn
I EU skal man forholde sig til behandling af børn i forbindelse med alle nye lægemidler. Generelt set skal de testes på børn. Der kan dispenseres fra dette krav, hvis sygdommen kun ses hos ældre mennesker eller voksne, eller hvis det nye lægemiddel sandsynligvis vil være virkningsløst eller være forbundet med risici hos alle eller nogle børn. Hvis der ikke gives dispensation, skal virksomheden udarbejde en undersøgelsesplan for børn – medmindre den får udsættelse. I så fald kan undersøgelsesplanen udarbejdes senere. Dette afsnit skal indeholde en kopi af dispensationen eller beslutninger om undersøgelsesplanen vedrørende børn (herunder eventuelle udsættelser).
Hvordan samles dossieret?
I de fleste tilfælde er det ikke længere muligt at indsende materiale på papir. Det betyder, at al dokumentationen i de fem FTD-moduler skal være i det elektroniske standardformat: eCTD. eCTD er ikke bare en samling PDF-dokumenter eller én enorm PDF-fil. eCTD er derimod en standard, som udførligt beskriver den mappe- og filstruktur, der skal følges, således at virksomheden og lægemiddelmyndighederne nemt kan navigere i dossieret, ligesom i et almindeligt computerbibliotek.
Hvordan er indsendelsesprocessen?
Ansøgeren skal omhyggeligt overveje alle logistiske spørgsmål og myndighedskrav forud for indsendelsen. Dette omfatter valg af den markedsføringstilladelsesprocedure, der skal følges: den centrale procedure (CP), den gensidige anerkendelsesprocedure (MRP – Mutual Recognition Procedure), den decentrale procedure (DCP) eller den nationale procedure (NP).
-
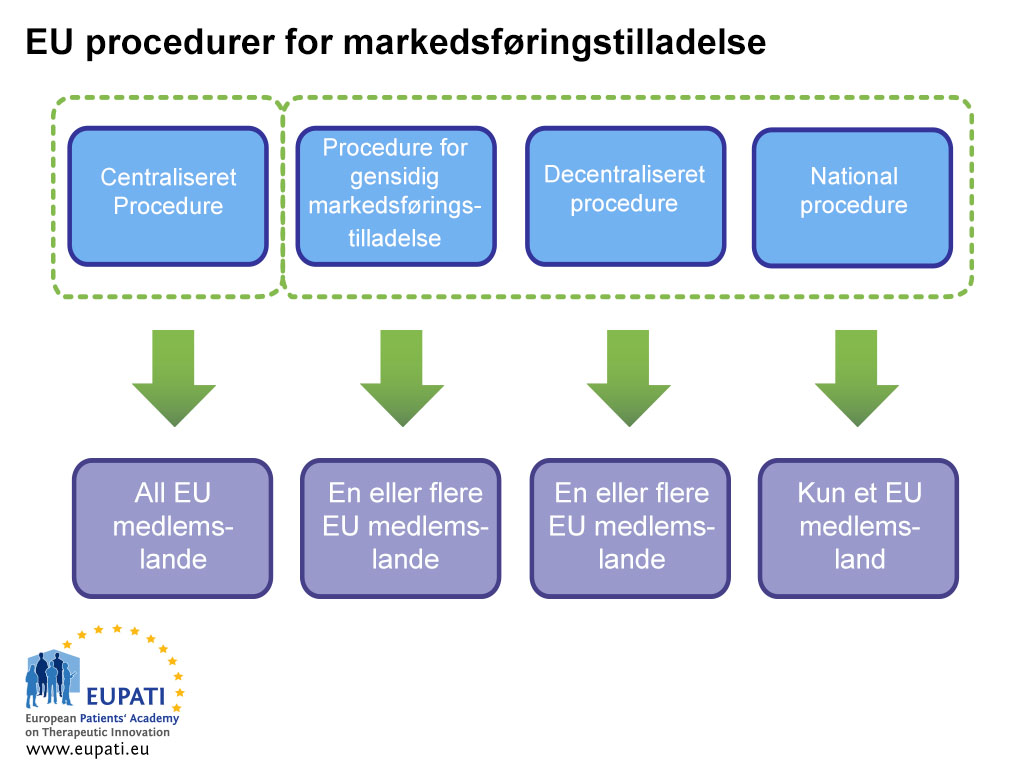
- Forskellige aktører er involveret I at give markedsføringstilladelse til et lægemiddel afhængigt af hvilken procedure sponsoren vælger (eller forpligtet til) at følge.
På EMA's hjemmeside findes der svar på mange spørgsmål vedrørende ansøgninger via CP.
Møder før indsendelsen
Møder før indsendelse af ansøgningen mellem virksomheden og personale fra lægemiddelmyndighederne finder normalt sted seks til syv måneder før datoen for indsendelse. Møderne planlægges sådan, at virksomheden kan søge yderligere oplysninger og vejledning, inden ansøgningsdossieret færdiggøres.
Ved MAA'er, der følger CP, mødes projektteamet fra virksomheden med det team fra EMA, som har til opgave at vurdere ansøgningen. Ved MRP, DCP eller NP er det muligt og ligeledes nyttigt at afholde møder med de relevante nationale kompetente myndigheder.
Indsendelse af ansøgning om markedsføringstilladelse (MAA)
Ved CP kan indsendelse til EMA kun ske i eCTD-format, medmindre der gives dispensation. eCTD indsendes via en onlineportal.
Ved ansøgninger, der følger MRP, DCP eller NP, er situationen mere kompliceret. Disse ansøgninger kan involvere op til 31 forskellige organer. HMA-netværket (Heads of Medicines Agencies network, et samarbejde mellem alle medlemsstater) tilbyder nu en løsning, der svarer til EMA's: CESP (Common European Submission Platform). Ved brug af CESP behøver virksomheden kun at uploade et dossier til systemet én gang. Alle involverede medlemsstater kan derefter hente det indsendte fra CESP-databasen. Platformen giver også mulighed for kommunikation mellem myndigheder og ansøgeren.
Valideringsfasen
Når EMA eller den nationale kompetente myndighed modtager den indsendte MAA, valideres dossieret først for at sikre, at al nødvendig dokumentation er med. Hvis der er spørgsmål, får ansøgeren mulighed for at give de nødvendige svar og understøttende dokumentation. Når MAA'en er valideret, begynder vurderingen.
A2-5.11-V1.1