Регистрационное свидетельство
Введение
Для официального выхода на рынок Европейской экономической зоны (ЕЭЗ) медицинские препараты должны сопровождаться регистрационным свидетельством.1 Главной задачей регистрационного свидетельства является обеспечение быстрого доступа к безопасным, эффективным и высококачественным медицинским препаратам для всех граждан на всей территории ЕЭЗ. Необходимо отметить, что медицинское оборудование проходит процессы, которые отличаются от предназначенных для другой медицинской продукции, до того, как начинает продаваться на рынке.
Национальные компетентные органы, или, сокращенно, «контрольно-надзорные органы» — это национальные правительственные органы, на которые возлагается ответственность за оценку заявок на получение регистрационных свидетельств и выдачу регистрационных свидетельств на медицинские препараты, которые появляются на рынках соответствующих стран после проведения национальных и децентрализованных процедур или процедур взаимного признания. Европейское агентство по лекарственным средствам (European Medicines Agency, EMA) в Лондоне осуществляет координацию научной оценки заявок на европейские регистрационные свидетельства на медицинские продукты в соответствии с централизованной процедурой (ЦП). Оценка производится экспертами национального контрольно-надзорного органа. После вынесения положительной оценки Европейская комиссия выдает регистрационное свидетельство.
Процедуры выдачи регистрационного свидетельства
Существует два способа получения регистрационного свидетельства: посредством централизованной или нецентрализованной процедуры. Последний вариант включает децентрализованную процедуру (ДЦП), процедуру взаимного признания (ПВП) и национальную процедуру. В каждой системе действуют отдельные положения законодательства и отдельные виды требований к компетентным органам (Европейскому агентству по лекарственным средствам или национальным компетентным органам и владельцам регистрационных свидетельств.
Централизованная процедура (ЦП)
Большинство новых медицинских препаратов проходят через эту процедуру. Представители отрасли подают единую заявку в Европейское агентство по лекарственным средствам. Комитет EMA по лекарственным препаратам для медицинского применения (Committee for Human Medicinal Products, CHMP), в который входят по одному члену от каждой страны-члена ЕС и страны ЕЭЗ, а также пять ученых экспертов, выносит рекомендацию в адрес Европейской комиссии относительно утверждения медицинского препарата. При утверждении медицинского препарата таким образом лицензия на продажу медицинского препарата выдается во всех странах ЕС и ЕЭЗ. Этот процесс научной оценки занимает не более 210 активных дней, хотя этот отсчет может быть приостановлен (это называется «остановкой часов»).
Децентрализованная процедура (ДЦП) и процедура взаимного признания (ПВП)
При указанных процедурах заявки на регистрационные свидетельства подаются в нескольких странах-членах ЕС, при этом одна страна принимает на себя функции страны-поручителя. После успешного прохождения процедуры медицинский препарат утверждается для продажи в стране, которая является поручителем, и в других задействованных странах (заинтересованных странах-членах ЕС). Вышеуказанные процедуры основаны на принципе признания оценки, осуществлённой страной-поручителем, заинтересованными странами-членами ЕС. Большинство медицинских препаратов-дженериков утверждаются посредством ДЦП.
Национальная процедура
Независимые национальные процедуры применяются строго для медицинских препаратов, которые будут сертифицированы и поступят на рынок только одной страны-члена ЕС. Эта процедура в настоящее время редко применяется для новой продукции.
Выбор процедуры получения регистрационного свидетельства
Фармацевтическая компания, как правило, может выбирать, какую процедуру использовать. Это решение относится к деловой сфере, и на выбор определенной процедуры влияет множество причин. Централизованная процедура является обязательной для
- продуктов, полученных в ходе биотехнологических процессов,
- лекарственных препаратов для передовой терапии (например, генной терапии, соматической клеточной терапии или препаратов на основе тканевой инженерии),
- орфанных лекарственных препаратов;
- медицинских препаратов для лечения ВИЧ или СПИДа, рака, нейродегенеративных расстройств, автоиммунной дисфункции и других дисфункций иммунной системы, вирусных заболеваний и диабета.
ЦП также может использоваться в случае с медицинскими препаратами, которые представляют собой значительную терапевтическую, научную или техническую инновацию или защищают интересы здоровья людей, например, медицинские препараты для снижения веса.
Если компания планирует разрабатывать существующий медицинский препарат для получения регистрационного свидетельства на использование по другим показаниям, обычно ей необходимо подавать заявку на абсолютно новое регистрационное свидетельство.
#mla_gallery-1 { margin: auto; width: 100%; } #mla_gallery-1 .gallery-item { float: none; margin: 1.5%; display: inline-block; text-align: center; width: 97%; } #mla_gallery-1 .gallery-item .gallery-icon img { border: 2px solid #cfcfcf; } #mla_gallery-1 .gallery-caption { margin-left: 0; vertical-align: top; } /* see mla_gallery_shortcode() in media-library-assistant/includes/class-mla-shortcode-support.php */
-
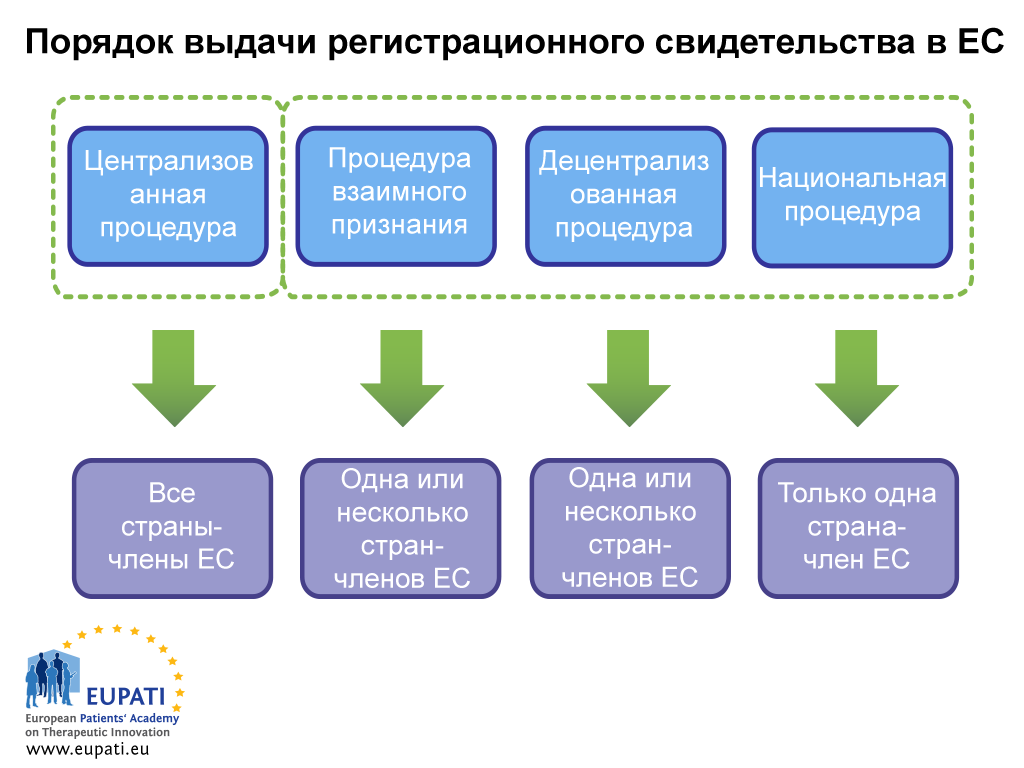
- зависимости от того, какую процедуру выбирает спонсор (или какой он вынужден следовать), процесс получения регистрационного свидетельства медицинского препарата может требовать участия разных сторон.
Научная оценка медицинских препаратов
Независимо от вида процедуры, для проведения оценки на получение регистрационного свидетельства на любой медицинский препарат требуется стандартное «досье». Общий технический документ (Common Technical Document, CTD) является упорядоченным форматом, признанным на международном уровне, которому необходимо следовать при оформлении заявок, предназначенных для подачи в контрольно-надзорные органы. Общий технический документ состоит из набора документов, в которых заявителю следует подтвердить надлежащее качество, безопасность и результативность медицинской продукции.
Отказ в выдаче регистрационного свидетельства
В выдаче регистрационного свидетельства на медицинский продукт будет отказано, если:
- баланс рисков и преимуществ рассматривается как неблагоприятный; или
- терапевтическая результативность недостаточно обоснована заявителем; или
- качественный и количественный состав не соответствует заявленному.
Действительность регистрационного свидетельства и дальнейшее сопровождение
Регистрационное свидетельство обновляется по истечении пяти лет на основе переоценки баланса преимуществ и рисков компетентным органом той страны, которая утверждает продукт. После обновления регистрационное свидетельство остается действительным на неопределенный период, если только компетентный орган не выносит, на разумном основании, связанном с фармакологическим надзором, решение о продлении на еще один дополнительный пятилетний срок.
Все владельцы регистрационных свидетельств обязаны предоставлять дальнейшие данные по безопасности и выполнять другие постмаркетинговые требования. После выхода на рынок медицинские препараты должны оставаться под наблюдением (фармакологическим надзором) с целью обеспечить обнаружение и оценку всех аспектов, которые могут повлиять на описание безопасности медицинского препарата, и предпринять необходимые меры.
В период нахождения медицинского препарата на рынке могут появляться новые данные относительно его результативности, безопасности или производства. В указанных случаях именно владелец регистрационного свидетельства обязан подать заявку на внесение изменений в регистрационного свидетельства в компетентные органы во всех странах, которые ранее утвердили эту медицинскую продукцию.
Участие пациентов в процессе выдачи регистрационного свидетельства
Участие пациентов в оформлении регистрационного свидетельства традиционно незначительно. Несколько компетентных органов, однако, рассматривают возможность задействования пациентов в процессе оформления регистрационного свидетельства — например, путем консультирования с клиентами и (или) задействования их в Комиссии по лекарственным препаратам для человека в составе Агентства по регулированию медицинских препаратов и изделий медицинского назначения (Medicines and Healthcare Products Regulatory Agency, MHRA) Великобритании. Пациенты уже принимают участие в обсуждении преимуществ и рисков в Европейском агентстве по лекарственным средствам в качестве членов Комитета по фармакологическому надзору, посредством участия в процедурах по научному обсуждению и (или) протоколу, а также во встречах Научной консультативной группы (Scientific Advisory Group, SAG). Кроме того, запущена пилотная инициатива, которая предполагает участие пациентов в Комитете по лекарственным препаратам для медицинского применения (СHMP).
Защитники прав пациентов также имеют возможность оказывать влияние на национальное законодательство и законодательство ЕС, относящееся к регистрационным свидетельствам.
Справочная литература
- The European Economic Area (EEA) unites the EU Member States and the three EEA EFTA States (Iceland, Liechtenstein, and Norway) into the internal market governed by the same basic rules. These rules aim to enable goods, services, capital, and persons to move freely about the EEA in an open and competitive environment, a concept referred to as the four freedoms.
Приложения
#mla_gallery-2 { margin: auto; width: 100%; } #mla_gallery-2 .gallery-item { float: none; margin: 1.5%; display: inline-block; text-align: center; width: 97%; } #mla_gallery-2 .gallery-item .gallery-icon img { border: 2px solid #cfcfcf; } #mla_gallery-2 .gallery-caption { margin-left: 0; vertical-align: top; } /* see mla_gallery_shortcode() in media-library-assistant/includes/class-mla-shortcode-support.php */
- Регистрационное свидетельство
Size: 772,128 bytes, Format: .pptx
Узнайте больше о процедурах получения регистрационного свидетельства медицинского препарата в Европейской экономической зоне.
A2-5.07-v1.1