Доклинические исследования: основные принципы
Введение
Доклинические исследования прежде всего направлены на определение варианта лечения, который имеет наибольшую вероятность успешного применения, оценку его безопасность и подведение аргументированной научной базы до перехода к этапу клинических исследований.
Кроме того, на этапе доклинических исследований исследуемое вещество должно быть приведено в соответствие с параметрами, не относящимися к медицине, включая определение прав интеллектуальной собственности и производство достаточного количества медицинского продукта для проведения клинических испытаний. Доклиническое исследование лекарственного препарата — это сложный регламентируемый процесс.
Основные понятия, ключевые определения, концепции
«Доклинические» или «предклинические»?
Термины «доклинический» и «предклинический» нередко используются в качестве тождественных.
Будучи исключительно важным аспектом на предклинических этапах разработки, доклинические исследования могут проводиться в любой момент жизненного цикла продукта, хотя их большая часть осуществляется как можно раньше во избежание неожиданностей в дальнейшем при проведении разработки.
Наряду с определением таких характеристик вещества, как фармакодинамика (какое действие препарат оказывает на организм), фармакокинетика (как организм реагирует на лекарственный препарат) и токсичность исследуемого вещества до его введения в человеческий организм, данные доклинических исследований используются для уточнения, объединения и добавления сведений к информации о безопасности продукта на этапе доклинических изысканий, в момент регистрации и на протяжении всего жизненного цикла медицинского продукта.
Компьютерное моделирование, исследования в лабораторных условиях, и исследования в естественных условиях
Исследования в рамках доклинических разработок проводятся в виде
- компьютерного моделирования: «выполняется на компьютере или посредством компьютерной симуляции», например, прогнозирование данных о токсичности продукта на основе сведений о его химической структуре, полученных на основе базы данных;
- исследований в лабораторных условиях (лат. «в стекле»): процесс осуществляется в контролируемых условиях вне живого организма, например, использование культур гепатоцитов (клеток печени) для изучения метаболизма;
- исследований в естественных условиях (лат. «в (на) живом»): эксперименты на целом живом организме в отличие от тканей или клеток, например, на животных, людях или растениях.
Какие основные аспекты стандарта по химическим свойствам, процессу производства и контролю качества (Chemistry, Manufacturing, Control, CMC) учитываются в доклинических исследованиях?
Любые исследования в ходе доклинических разработок требуют производства достаточного количества активного вещества:
- для доклинических исследований обычно требуется незначительное количество (от миллиграммов до граммов), в дальнейшем необходимо налаживать более масштабное производство для получения объемов, необходимых для клинических исследований, и впоследствии, после утверждения препарата, для его вывода на рынок.
- Для проведения исследований по нормам надлежащей лабораторной практики (GLP) требуются партии активного вещества, произведенные по нормам надлежащей производственной практики (GMP).
Некоторые ключевые стадии для соблюдения стандарта CMC на этапе доклинических исследований включают:
- определение дозы и способа введения;
- подробное описание физико-химических характеристик;
- испытания на стабильность и определение содержания примесей;
- разработка и оценка методов определения количественного содержания активного вещества в биологических жидкостях организма (кровь, плазма, моча) и изучение побочных эффектов;
- разработка прототипа препарата, который будет использоваться в клинических условиях.
Процесс доклинических исследований
План проведения доклинических исследований аналогичен плану проведения научного исследования. Доклинические исследования направлены на поддержание запланированной программы клинических разработок путем решения задач и получения ответа на вопросы, которые приведены ниже.
Задачи
После того как определен состав исследуемого препарата, в процессе доклинических разработок исследователи проводят специальные оценки и исследования, чтобы получить ответы на следующие вопросы:
- Действует ли препарат? оценка эффективности
- Как препарат будет доставляться и какова реакция организма? → составление описания препарата
- Безопасен ли препарат? → токсичность/безопасность
- Является ли производство экономически целесообразным и контролируемым?
Процесс доклинических исследований может происходить на протяжении всего жизненного цикла продукта, однако при этом чем раньше будут получены ответы на эти вопросы, тем скорее можно будет определить, какие пациенты получат наибольшую пользу от назначения этого препарата.
Управление проектом
Программа доклинических исследований является сложным процессом и требует навыков по управлению проектами, а также навыков общения при работе с командами, состоящими из специалистов, занимающихся различными направлениями. Группе, занятой в проекте, необходимо получить представление о плане клинических исследований для того, чтобы определить план доклинических исследований и связанных с ними видов работ.
Описание проекта обеспечивает основу для реализации стратегии доклинических исследований, определения целей, рисков, обязательств, контрольных показателей и принятия решений о продолжении или прекращении исследования. Реализация проекта согласно описанию способствует концентрации на ключевых критериях продукта, своевременному принятию решений о продолжении или прекращении исследования и сокращению рисков, связанных с проектом в целом (т.е. риска длительной разработки продукта, не являющегося полезным).
Руководство по проведению доклинических исследований
Разработкой препаратов занимаются многие участники фармацевтической отрасли, и все организации и учреждения следуют своим собственным правилам. Например, компании-производители руководствуются стандартными операционными процедурами (СОП). Наряду со стандартом «надлежащей клинической практики» используются также рекомендации, которые находятся на интернет-сайте Европейского агентства по лекарственным средствам (European Medicines Agency, EMA).
- Там находятся как рекомендации общего характера, так и более конкретные рекомендации по отдельным научным и техническим аспектам (например, отдельные инструкции по изучению токсичности).
- Для подачи заявки на разрешение на поставку этим рекомендациям необходимо следовать неукоснительно; любое отклонение от нормы должно быть оправдано.
Данные предоставляются в соответствии с форматом, приведенным в «общем техническом документе» (CTD), который утвержден Международной конференцией по гармонизации технических требований к регистрации фармацевтических продуктов, предназначенных для применения человеком (International Conference on Harmonisation, ICH). Соглашение об объединении всей информации, касающейся качества, безопасности и эффективности, в этом общем формате (CTD) стало революционным прорывом в процессе организации контрольно-надзорных мероприятий и позволило гармонизировать подачу сведений в электронной форме, что, в свою очередь, обеспечило введение надлежащих процедур контроля. Для субъектов отрасли исчезла необходимость подачи информации в разных форматах в разные контрольно-надзорные органы (Международная конференция по гармонизации объединяет контрольно-надзорные органы и фармацевтические компании Европы, Японии и США для целей обсуждения различных научных и технических аспектов регистрации лекарственных препаратов).
-
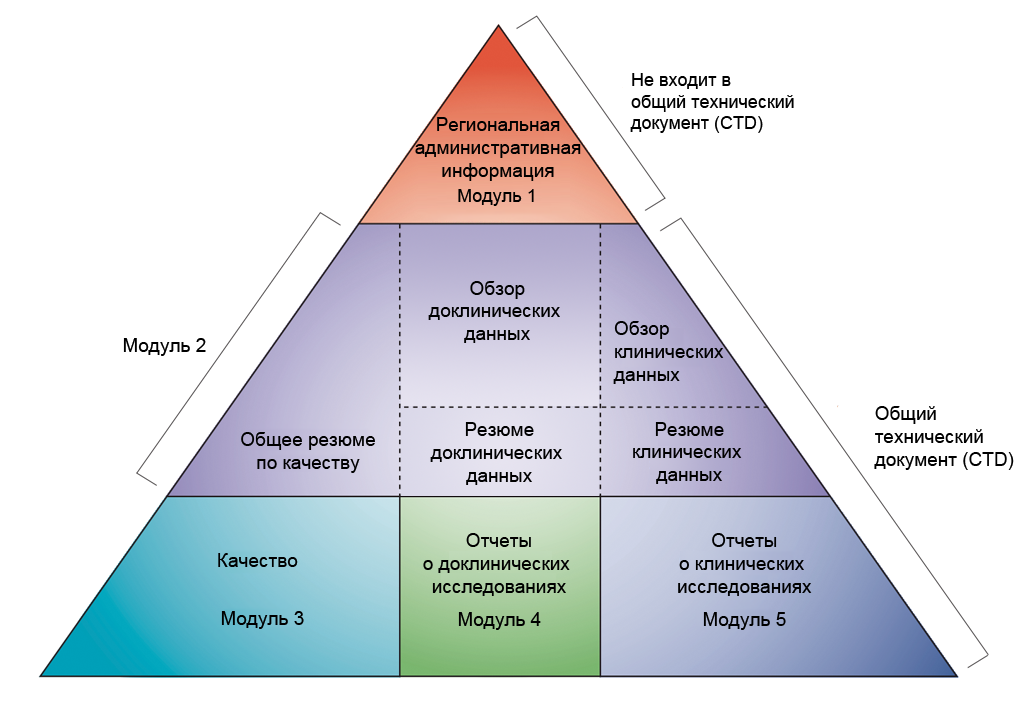
- Доклиническая разработка в модулях CTD. По материалам ICH CTD (см. ссылку 1)
Общий технический документ (CTD) состоит из пяти модулей (см. рисунок выше). С июля 2003 г. общий технический документ представляет собой обязательный формат для всех заявок для получения разрешения на поставку в Европе и Японии и настоятельно рекомендуется в качестве оптимального формата для подачи заявки на регистрацию нового препарата в Управление США по надзору за качеством пищевых продуктов и лекарственных средств (US Food and Drug Administration, FDA).
Резюме
Этап доклинических исследований очень важен, поскольку потенциальные проблемы необходимо обнаружить именно в этот период, прежде чем начнется этап клинических исследований данного препарата.
Для допуска исследуемого вещества на этап клинических испытаний необходимо
- проводить оценку безопасности доклинических исследований согласно условиям надлежащей лабораторной практики (GLP),
- осуществлять производство с соблюдением надлежащего контроля качества,
- фиксировать данные и описание процесса в формате общего технического документа и создать основу для этапа клинических исследований.
Растет тенденция к разработке лекарственных препаратов с помощью компьютерного моделирования, а также с использованием методов биоинформатики при моделировании и прогнозировании.
Основными контрольно-надзорными органами (стран Европы, США и Канады) ведется постоянная работа по упорядочению рекомендаций по проведению доклинических исследований с акцентом на безопасность и качество: Международная конференция по гармонизации публикует подробные рекомендации для фармацевтической отрасли по аналогии с изданиями Европейского агентства по лекарственным средствам (EMA) и Управления США по надзору за качеством пищевых продуктов и лекарственных средств (FDA).
Дополнительные источники
- European Medicines Agency (2007a). Guideline on strategies to identify and mitigate risks for first-in human trials with investigational medicinal products. London: European Medicines Agency. Retrieved 25 June, 2015, from http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002988.pdf
- European Medicines Agency (2007b). Guideline on requirements for first-in-man clinical trials for potential high-risk medicinal Retrieved 25 June, 2015, from http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002989.pdf
- European Medicines Agency (2009). ICH guideline M3(R2) on non-clinical safety studies for the conduct of human clinical trials and marketing authorisation for pharmaceuticals. London: European Medicines Agency. Retrieved 25 June, 2015, from http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002720.pdf
- European Medicines Agency (2011). Committee for medicinal products for human use (CHMP) ICH guidelines S6 (R1) – preclinical safety evaluation of biotechnology-derived pharmaceuticals. London: European Medicines Agency. Retrieved 25 June, 2015, from http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002828.pdf
[glossary_exclude]Справочная литература
- Image reproduced from ICH (2015). M4: The Common Technical Document. Retrieved 11 July, 2021, from https://www.ich.org/page/ctd
- European Medicines Agency (2009). ICH guideline M3(R2) on non-clinical safety studies for the conduct of human clinical trials and marketing authorisation for pharmaceuticals. London: European Medicines Agency. Retrieved 25 June, 2015, from http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002720.pdf[/glossary_exclude]